R sample code
Brendon Smith
br3ndonland
Source code on
GitHub

Data structures
Data frame
Example from Cookbook for R
Sample data frame
sample_data <-
data.frame (
id = 1:3,
weight = c(20, 27, 24),
size = c('small', 'large', 'medium')
)
sample_data## id weight size
## 1 1 20 small
## 2 2 27 large
## 3 3 24 mediumSort data frame by column number
## id size weight
## 1 1 small 20
## 2 2 large 27
## 3 3 medium 24Example from Stack Overflow
Sample data frame
dd <-
data.frame(
b = factor(
c('Hi', 'Med', 'Hi', 'Low'),
levels = c('Low', 'Med', 'Hi'),
ordered = TRUE
),
x = c('A', 'D', 'A', 'C'),
y = c(8, 3, 9, 9),
z = c(1, 1, 1, 2)
)
dd## b x y z
## 1 Hi A 8 1
## 2 Med D 3 1
## 3 Hi A 9 1
## 4 Low C 9 2Sort data frame by column
Sorting the frame by column z (descending) then by column b(ascending)
## b x y z
## 4 Low C 9 2
## 2 Med D 3 1
## 1 Hi A 8 1
## 3 Hi A 9 1Sort data frame by column index
Edit some 2+ years later: It was just asked how to do this by column index. The answer is to simply pass the desired sorting column(s) to the order() function rather than using the name of the column (and with() for easier/more direct access).
## b x y z
## 4 Low C 9 2
## 2 Med D 3 1
## 1 Hi A 8 1
## 3 Hi A 9 1Statistical models
T-test
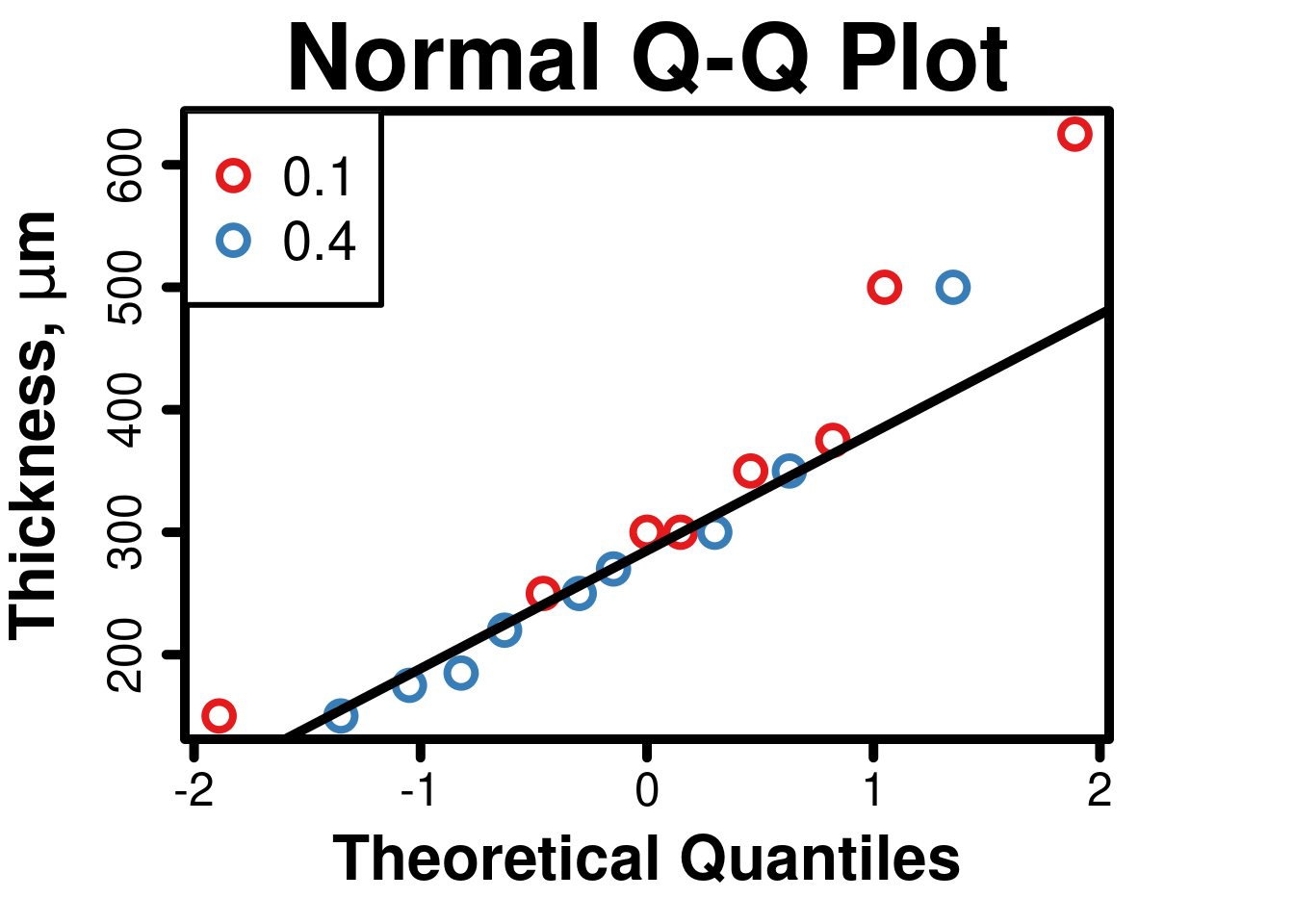
Atheroma thickness
Atheroma thickness data from Smith BW J Food Res 2013 (doi: 10.5539/jfr.v2n1p168).
Import dataset
jfr2013_atheroma <- read.table('data/t_test_jfr2013_atheroma.csv',
header = TRUE,
sep = ',')
attach(jfr2013_atheroma)
names(jfr2013_atheroma)## [1] "diet" "thickness"Perform t-test
- The default assumes unequal variance and applies the Satterthwaite/Welch df modification.
- var.equal = TRUE is used to specify equal variances and a pooled variance estimate.
t.test(
jfr2013_atheroma$thickness ~ jfr2013_atheroma$diet,
paired = FALSE,
alternative = 'two.sided',
var.equal = TRUE,
conf.level = 0.95
)##
## Two Sample t-test
##
## data: jfr2013_atheroma$thickness by jfr2013_atheroma$diet
## t = 1.4364, df = 15, p-value = 0.1714
## alternative hypothesis: true difference in means between group 0.1 and group 0.4 is not equal to 0
## 95 percent confidence interval:
## -43.34361 222.51028
## sample estimates:
## mean in group 0.1 mean in group 0.4
## 356.2500 266.6667Test assumptions
The t test uses raw data, not residuals, so assumptions are tested using raw data.
Normality
- The RColorBrewer package is loaded for additional colors, and a
palette is added to R. Use brackets after vector name for specific
colors when creating plots (for example:
col=set1[4:14]). - Legend can also be manually positioned with xy coordinates (numbers from 0,0 at bottom left of whole plot to 1,1 at top right)
##
## Shapiro-Wilk normality test
##
## data: thickness
## W = 0.91231, p-value = 0.1094# Normal Quantile-Quantile Plot
# Set graphical parameters
par(
cex = 1,
cex.main = 3,
cex.lab = 2,
font.lab = 2,
cex.axis = 1.5,
lwd = 5,
lend = 0,
ljoin = 0,
tcl = -0.5,
pch = 1,
mar = c(5, 5, 3, 5)
)
# Load RColorBrewer and set palette
library(RColorBrewer)
set1 <- brewer.pal(8, 'Set1')
# Create vector for categories
categories <- factor(jfr2013_atheroma$diet)
# Create plot
qqnorm(
jfr2013_atheroma$thickness,
ylab = expression(bold(paste(
'Thickness, ', mu, 'm'
))),
axes = FALSE,
col = set1[categories],
lwd = 4,
cex = 2
)
# Add normal line
qqline(jfr2013_atheroma$thickness, lwd = 5)
# Add border to complete axes
box(which = 'plot', bty = 'o', lwd = 5)
axis(1, lwd = 5, tcl = -0.5) # x axis
axis(2, lwd = 5, tcl = -0.5) # y axis
# Add legend
legend(
'topleft',
legend = levels(categories),
box.lwd = 3,
pch = 1.5,
cex = 1.75,
pt.cex = 2,
pt.lwd = 4,
col = set1
)
Equality of variances
##
## F test to compare two variances
##
## data: jfr2013_atheroma$thickness by jfr2013_atheroma$diet
## F = 1.8725, num df = 7, denom df = 8, p-value = 0.3983
## alternative hypothesis: true ratio of variances is not equal to 1
## 95 percent confidence interval:
## 0.4134808 9.1738855
## sample estimates:
## ratio of variances
## 1.872473Boxplot
- Parameters (
par): Graphical parameters need to be set again because of code chunk separation in RMarkdown. Normally they would only be set once per R session. - X axis: The default axes will usually detect
categorical data and label axes accordingly, but categories will need to
be specified for axes created separately. It is easiest to create a
separate vector with the categories, and then reference it when creating
the plot. Labels can also be specified with
labels=c('group1', 'group2'). - Y axis: Note that it does not always start at zero, but encompasses the data range with some padding.
# Set graphical parameters
par(
cex = 1,
cex.main = 3,
cex.lab = 2,
font.lab = 2,
cex.axis = 1.5,
lwd = 5,
lend = 0,
ljoin = 0,
tcl = -0.5,
pch = 1,
mar = c(5, 5, 3, 5)
)
# Create boxplot
boxplot(
jfr2013_atheroma$thickness ~ jfr2013_atheroma$diet,
main = 'Atheroma Thickness',
xlab = 'Diet',
ylab = expression(bold(paste(
'Thickness, ', mu, 'm'
))),
axes = FALSE,
lwd = 4,
medlwd = 7,
outlwd = 4,
cex = 2,
col = set1
)
# Add border to complete axes
box(which = 'plot', bty = 'l', lwd = 5)
# Create vector with means of each group
means <- aggregate(thickness ~ diet, jfr2013_atheroma, mean)
# Add points for group means to boxplot
points(
means$thickness,
cex = 2,
pch = 23,
bg = 0,
lwd = 4
)
# X axis
axis(
1,
lwd = 5,
tcl = -0.5,
labels = levels(categories),
at = c(1:2)
)
# Y axis
axis(2, lwd = 5, tcl = -0.5)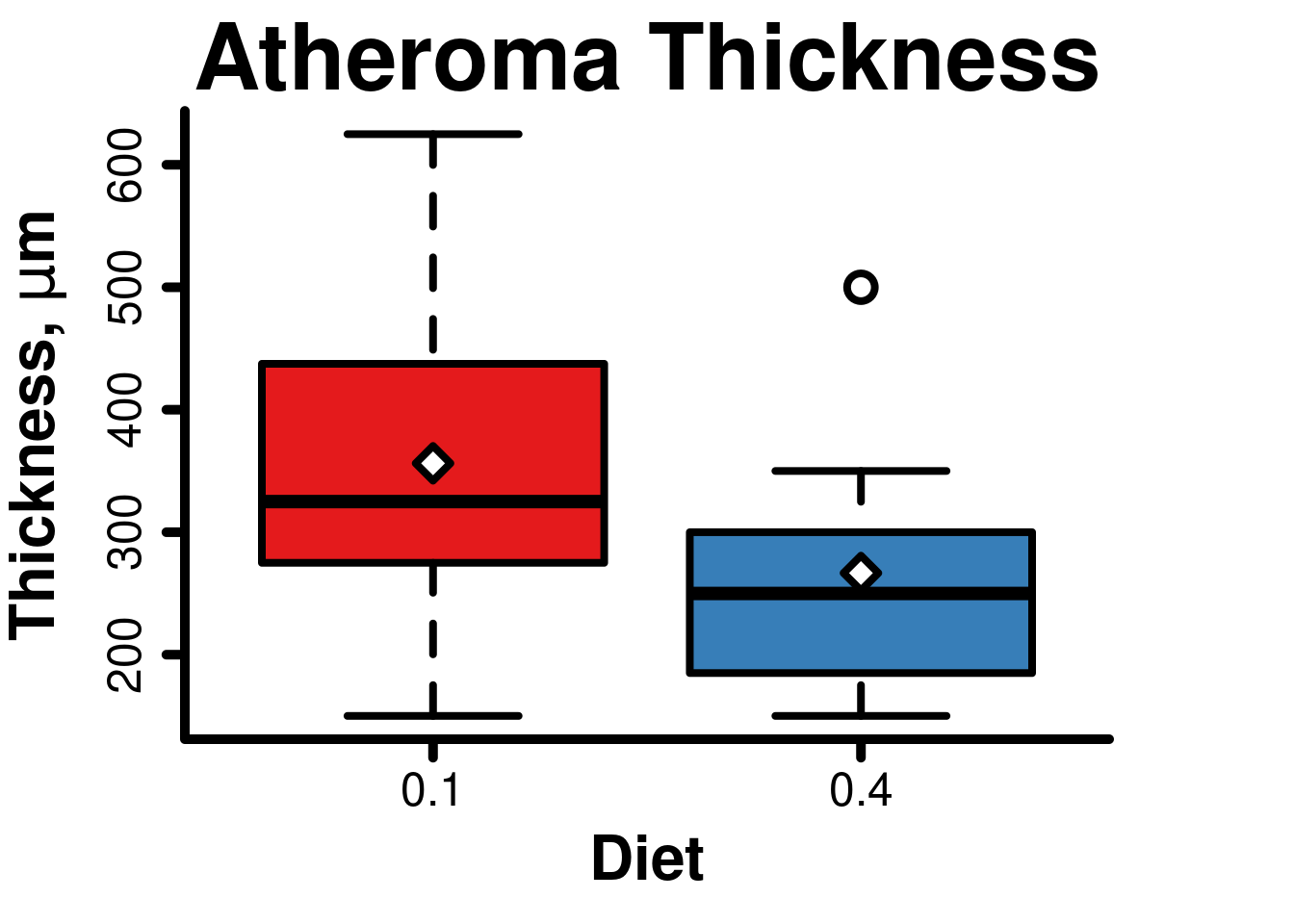
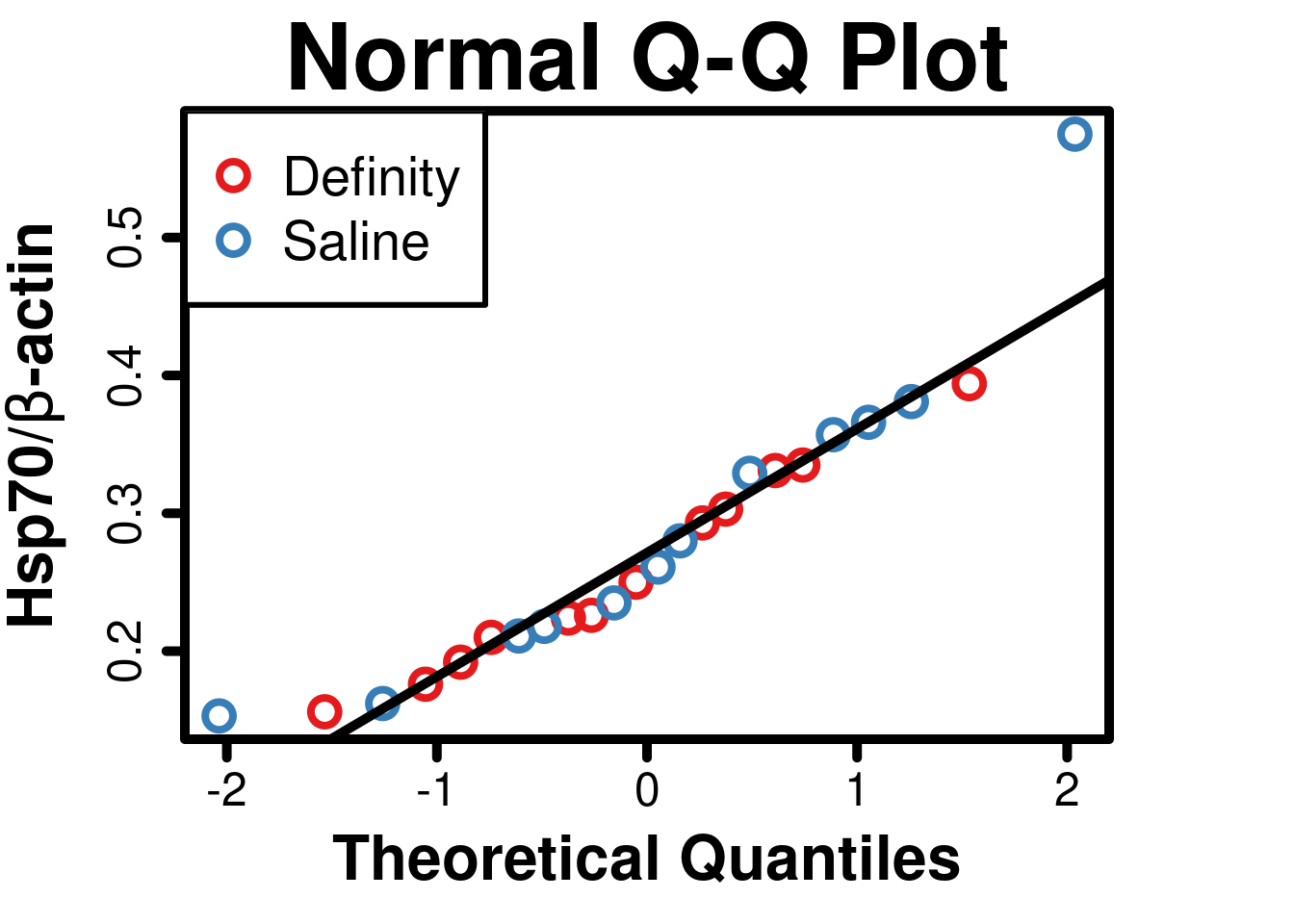
Aorta Hsp70
Aorta Hsp70 data from Smith BW J Ultrasound Med 2015 (doi: 10.7863/ultra.34.7.1209).
Import dataset
AortaHsp70 <- read.table('data/t_test_jum2015_hsp70.csv',
header = TRUE,
sep = ',')
attach(AortaHsp70)
names(AortaHsp70)## [1] "exposure" "Hsp70"Perform t-test
- The default assumes unequal variance and applies the Satterthwaite/Welch df modification.
- var.equal = TRUE is used to specify equal variances and a pooled variance estimate.
t.test(
AortaHsp70$Hsp70 ~ AortaHsp70$exposure,
paired = FALSE,
alternative = 'two.sided',
var.equal = TRUE,
conf.level = 0.95
)##
## Two Sample t-test
##
## data: AortaHsp70$Hsp70 by AortaHsp70$exposure
## t = -0.91363, df = 22, p-value = 0.3708
## alternative hypothesis: true difference in means between group Definity and group Saline is not equal to 0
## 95 percent confidence interval:
## -0.11935215 0.04635215
## sample estimates:
## mean in group Definity mean in group Saline
## 0.2575 0.2940Test assumptions
The t test uses raw data, not residuals, so assumptions are tested using raw data.
Normality
- The RColorBrewer package is loaded for additional colors, and a
palette is added to R. Use brackets after vector name for specific
colors when creating plots (for example:
col=set1[4:14]). - Legend can also be manually positioned with xy coordinates (numbers from 0,0 at bottom left of whole plot to 1,1 at top right)
##
## Shapiro-Wilk normality test
##
## data: AortaHsp70$Hsp70
## W = 0.90895, p-value = 0.03347# Normal Quantile-Quantile Plot
# Set graphical parameters
par(
cex = 1,
cex.main = 3,
cex.lab = 2,
font.lab = 2,
cex.axis = 1.5,
lwd = 5,
lend = 0,
ljoin = 0,
tcl = -0.5,
pch = 1,
mar = c(5, 5, 3, 5)
)
# Load RColorBrewer and set palette
library(RColorBrewer)
set1 <- brewer.pal(8, 'Set1')
# Create vector for categories
categories <- factor(AortaHsp70$exposure)
# Create plot
qqnorm(
Hsp70,
ylab = expression(bold(paste(
'Hsp70/', beta, '-actin'
))),
axes = FALSE,
col = set1[categories],
lwd = 4,
cex = 2
)
# Add normal line
qqline(AortaHsp70$Hsp70, lwd = 5)
# Add border to complete axes
box(which = 'plot', bty = 'o', lwd = 5)
axis(1, lwd = 5, tcl = -0.5) # x axis
axis(2, lwd = 5, tcl = -0.5) # y axis
# Add legend
legend(
'topleft',
legend = levels(categories),
box.lwd = 3,
pch = 1.5,
cex = 1.75,
pt.cex = 2,
pt.lwd = 4,
col = set1
)
Equality of variances
##
## F test to compare two variances
##
## data: AortaHsp70$Hsp70 by AortaHsp70$exposure
## F = 0.38929, num df = 11, denom df = 11, p-value = 0.1328
## alternative hypothesis: true ratio of variances is not equal to 1
## 95 percent confidence interval:
## 0.1120669 1.3522650
## sample estimates:
## ratio of variances
## 0.3892868Boxplot
- Parameters (
par): Graphical parameters need to be set again because of code chunk separation in RMarkdown. Normally they would only be set once per R session. - X axis: The default axes will usually detect
categorical data and label axes accordingly, but categories will need to
be specified for axes created separately. It is easiest to create a
separate vector with the categories, and then reference it when creating
the plot. Labels can also be specified with
labels=c('group1', 'group2'). - Y axis: Note that it does not always start at zero, but encompasses the data range with some padding.
# Set graphical parameters
par(
cex = 1,
cex.main = 3,
cex.lab = 2,
font.lab = 2,
cex.axis = 1.5,
lwd = 5,
lend = 0,
ljoin = 0,
tcl = -0.5,
pch = 1,
mar = c(5, 5, 3, 5)
)
# Create boxplot
boxplot(
AortaHsp70$Hsp70 ~ AortaHsp70$exposure,
main = 'Aorta Hsp70',
xlab = 'Exposure',
ylab = expression(bold(paste(
'Hsp70/', beta, '-actin'
))),
axes = FALSE,
lwd = 4,
medlwd = 7,
outlwd = 4,
cex = 2,
col = set1
)
# Add border to complete axes
box(which = 'plot', bty = 'l', lwd = 5)
# Create vector with means of each group
means <- aggregate(Hsp70 ~ exposure, AortaHsp70, mean)
# Add points for group means to boxplot
points(
means$Hsp70,
cex = 2,
pch = 23,
bg = 0,
lwd = 4
)
# X axis
axis(
1,
lwd = 5,
tcl = -0.5,
labels = levels(categories),
at = c(1:2)
)
# Y axis
axis(2, lwd = 5, tcl = -0.5)
Linear regression
Simple linear regression aztcpm
aztcpm dataset from University of Illinois statistics
course PATH
591 lab 02 and PATH
591 lecture 04 A
Intro
These data are from an in-vitro study of the effect of AZT dose on DNA replication (CPM) for FeLV (feline leukemia virus). (The experimental unit is the cell culture sample). For the linear regression analysis, the dependent variable is CPM (counts per minute, indicating quantity of Feline Leukemia Virus DNA in a cell culture) and the independent (predictor) variable is DOSE (dose of AZT).
Import dataset
## [1] "DOSE" "CPM"Perform regression diagnostics and test assumptions
##
## Call:
## lm(formula = CPM ~ DOSE)
##
## Residuals:
## Min 1Q Median 3Q Max
## -8712 -5708 -2483 3224 21640
##
## Coefficients:
## Estimate Std. Error t value Pr(>|t|)
## (Intercept) 39810.58 1711.46 23.261 < 2e-16 ***
## DOSE -813.61 89.45 -9.096 2.93e-10 ***
## ---
## Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
##
## Residual standard error: 8215 on 31 degrees of freedom
## Multiple R-squared: 0.7274, Adjusted R-squared: 0.7186
## F-statistic: 82.73 on 1 and 31 DF, p-value: 2.931e-10
##
##
## ASSESSMENT OF THE LINEAR MODEL ASSUMPTIONS
## USING THE GLOBAL TEST ON 4 DEGREES-OF-FREEDOM:
## Level of Significance = 0.05
##
## Call:
## gvlma(x = lm1)
##
## Value p-value Decision
## Global Stat 32.863 1.274e-06 Assumptions NOT satisfied!
## Skewness 9.168 2.463e-03 Assumptions NOT satisfied!
## Kurtosis 1.413 2.346e-01 Assumptions acceptable.
## Link Function 12.218 4.733e-04 Assumptions NOT satisfied!
## Heteroscedasticity 10.064 1.512e-03 Assumptions NOT satisfied!##
## Shapiro-Wilk normality test
##
## data: rstandard(lm1)
## W = 0.85782, p-value = 0.0005083## Warning in cor.test.default(fitted(lm1), abs(rstandard(lm1)), method =
## "spearman"): Cannot compute exact p-value with ties##
## Spearman's rank correlation rho
##
## data: fitted(lm1) and abs(rstandard(lm1))
## S = 6143.9, p-value = 0.8827
## alternative hypothesis: true rho is not equal to 0
## sample estimates:
## rho
## -0.02671635## Loading required package: carData## lag Autocorrelation D-W Statistic p-value
## 1 0.797716 0.1772224 0
## Alternative hypothesis: rho != 0## No Studentized residuals with Bonferroni p < 0.05
## Largest |rstudent|:
## rstudent unadjusted p-value Bonferroni p
## 33 3.027239 0.0050324 0.16607Generate plots
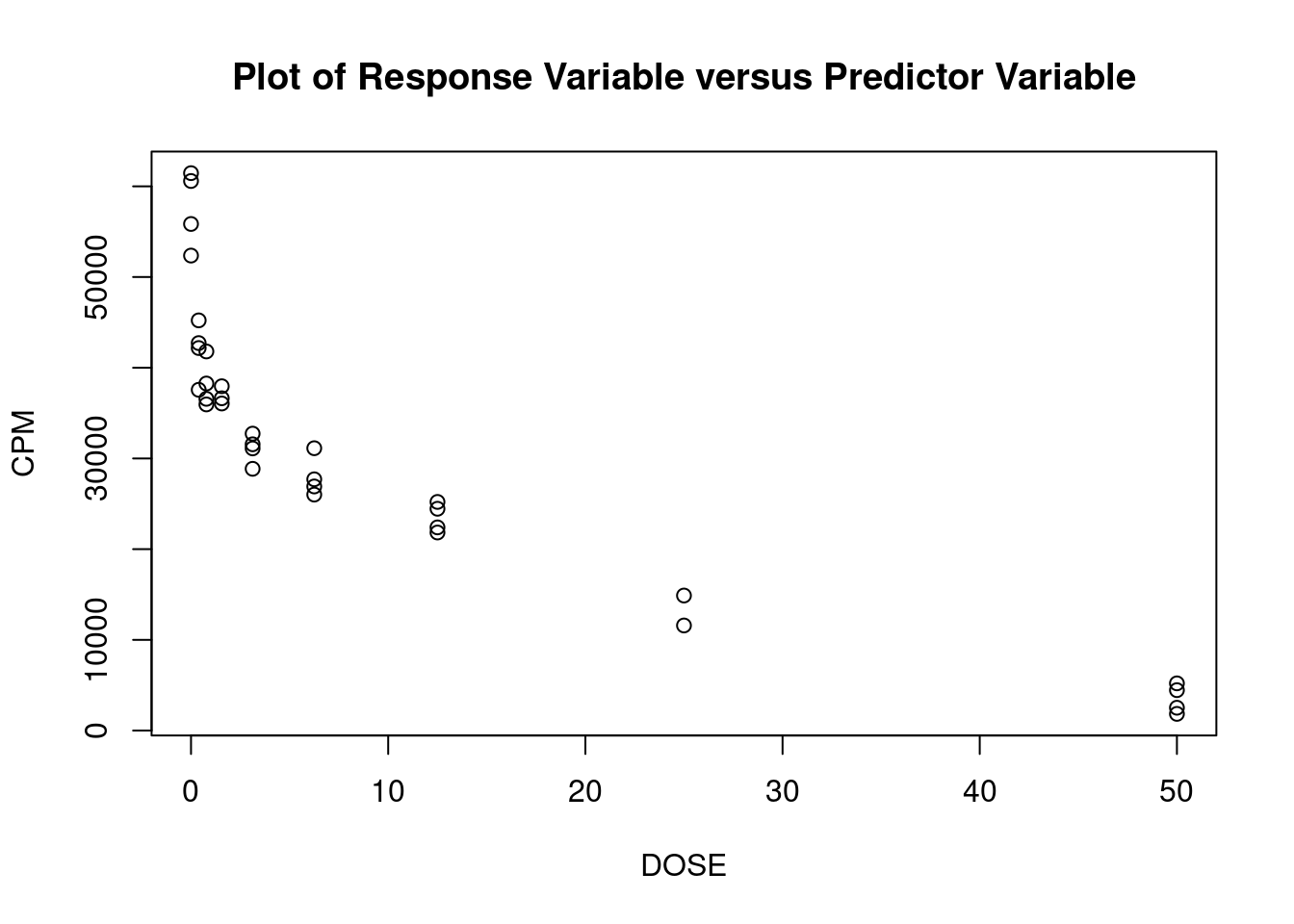
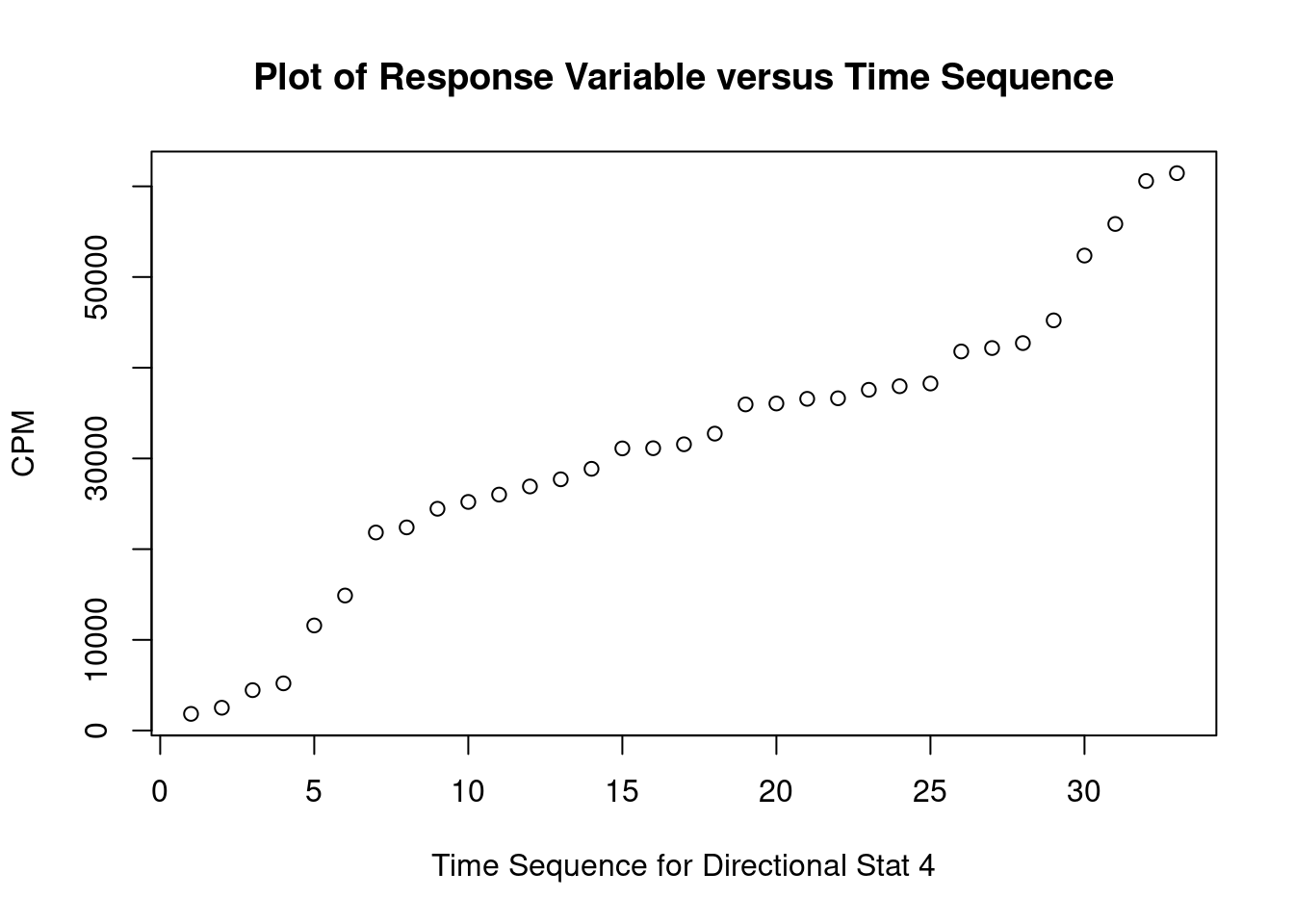
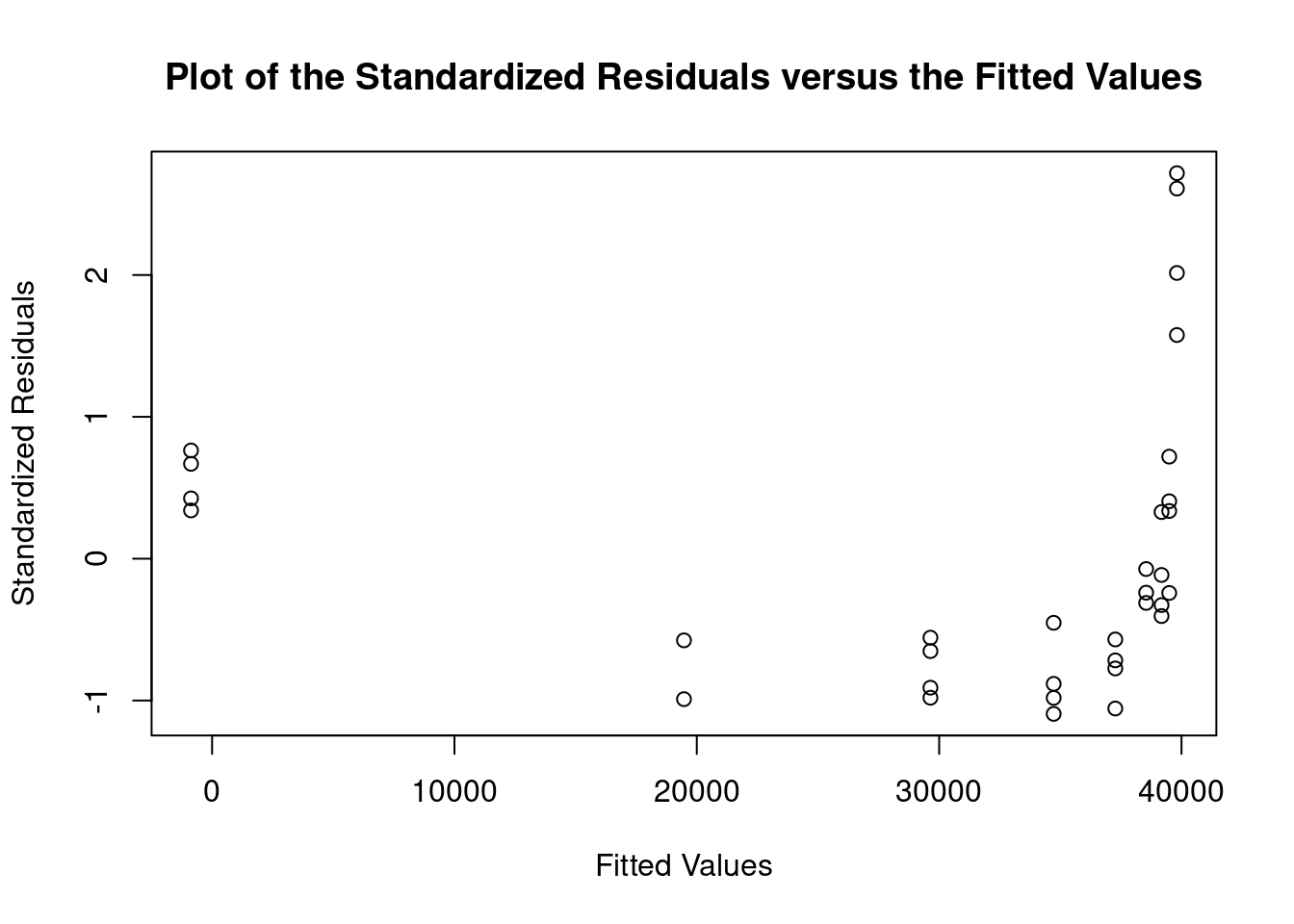
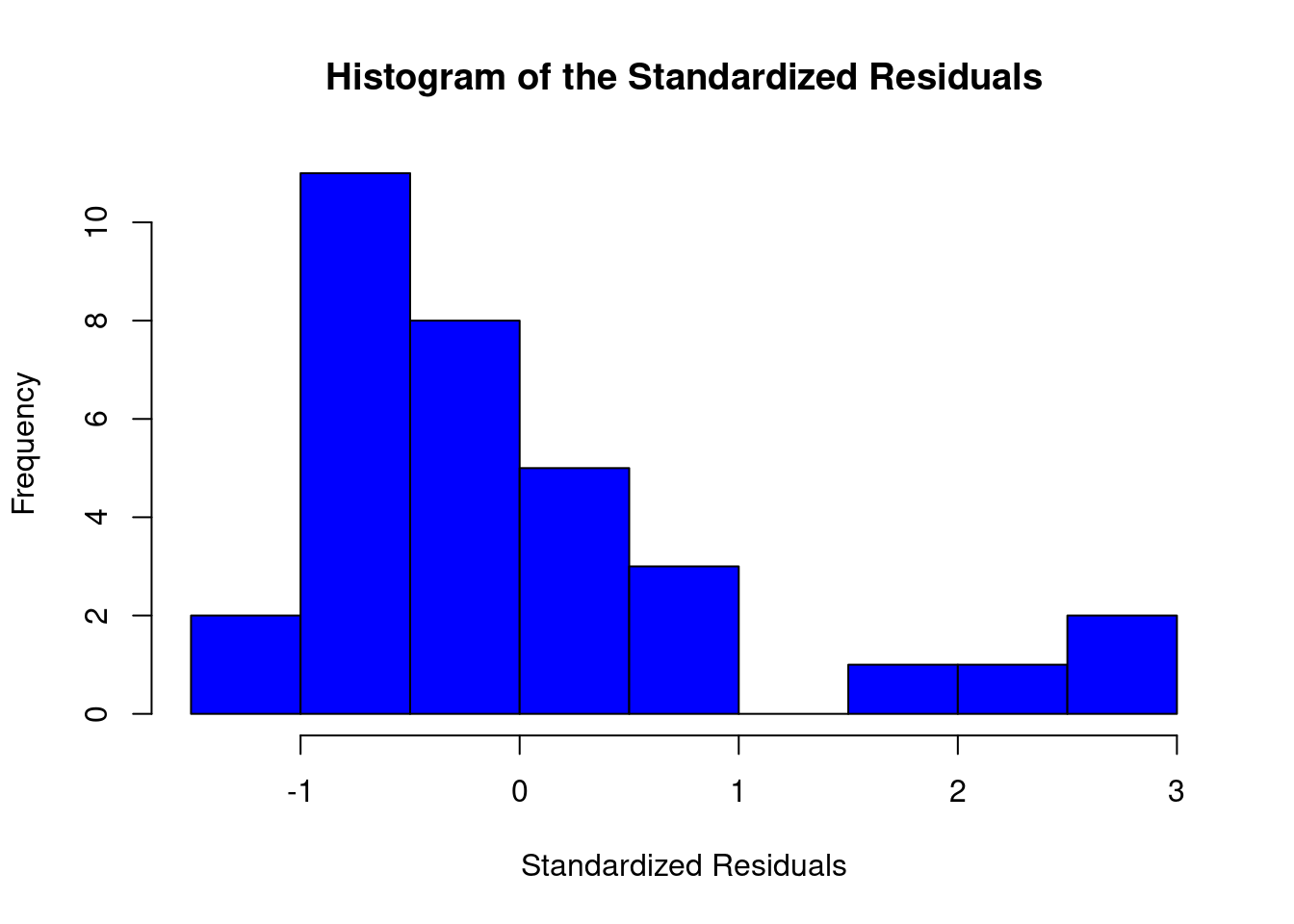
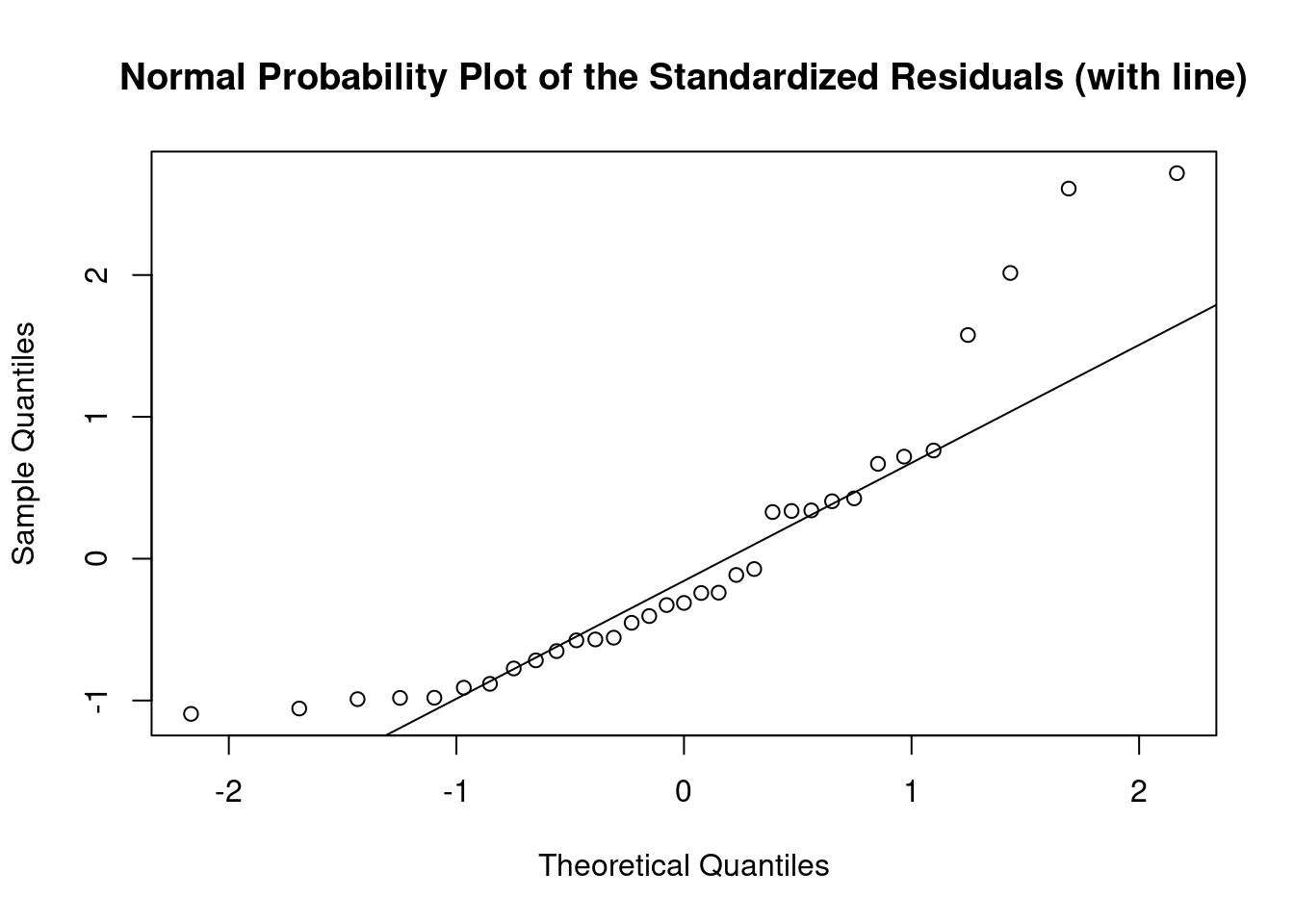
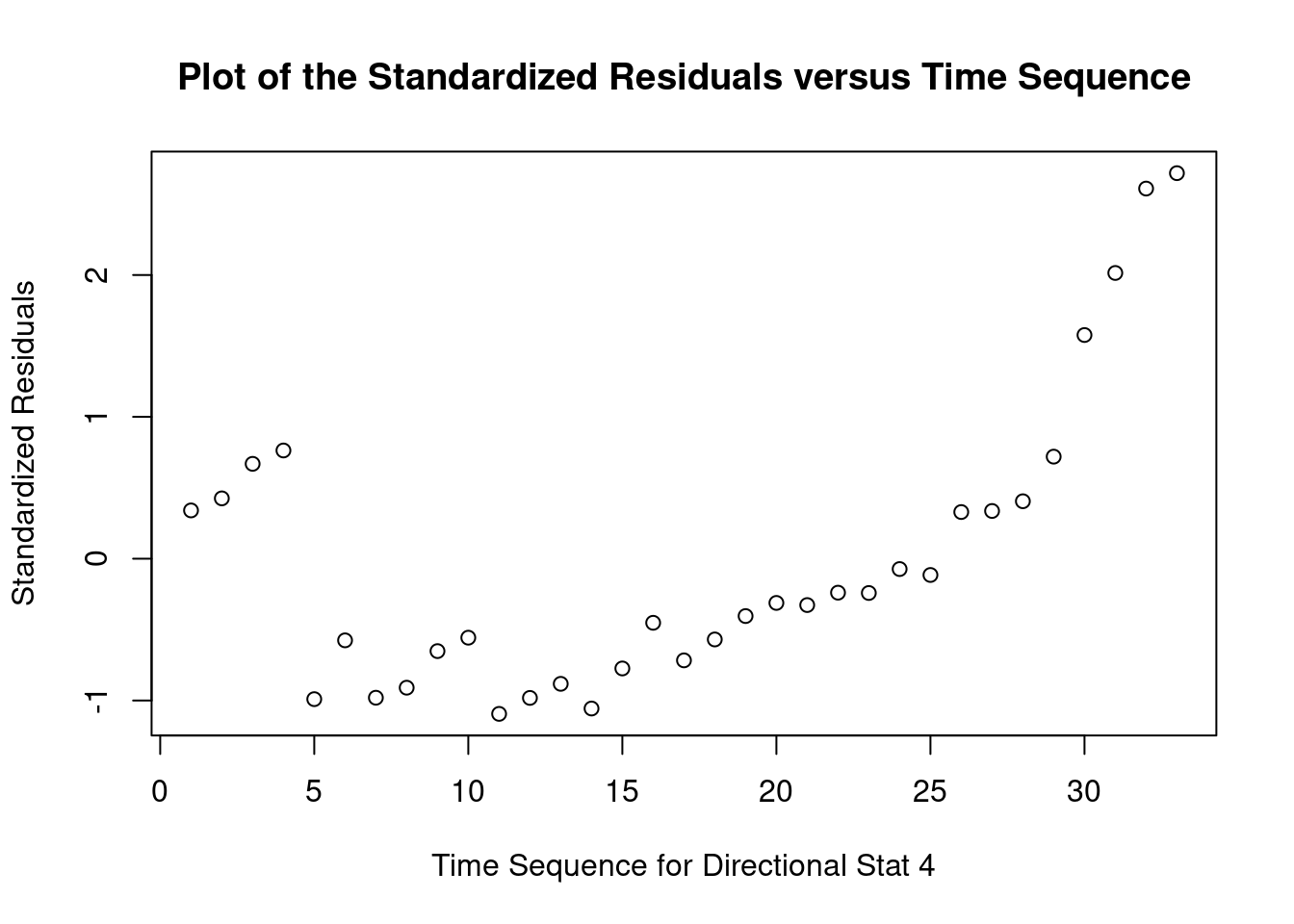
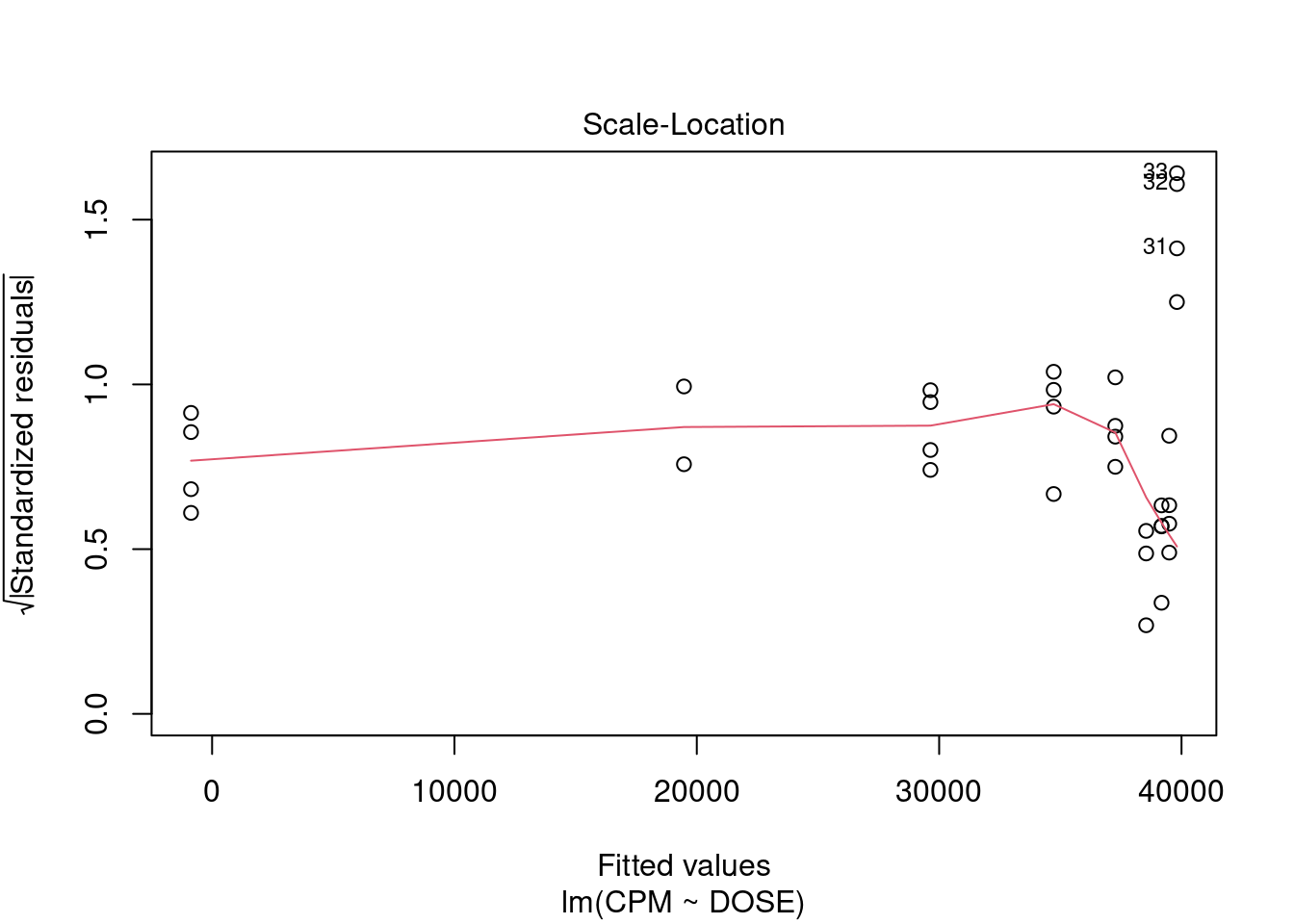
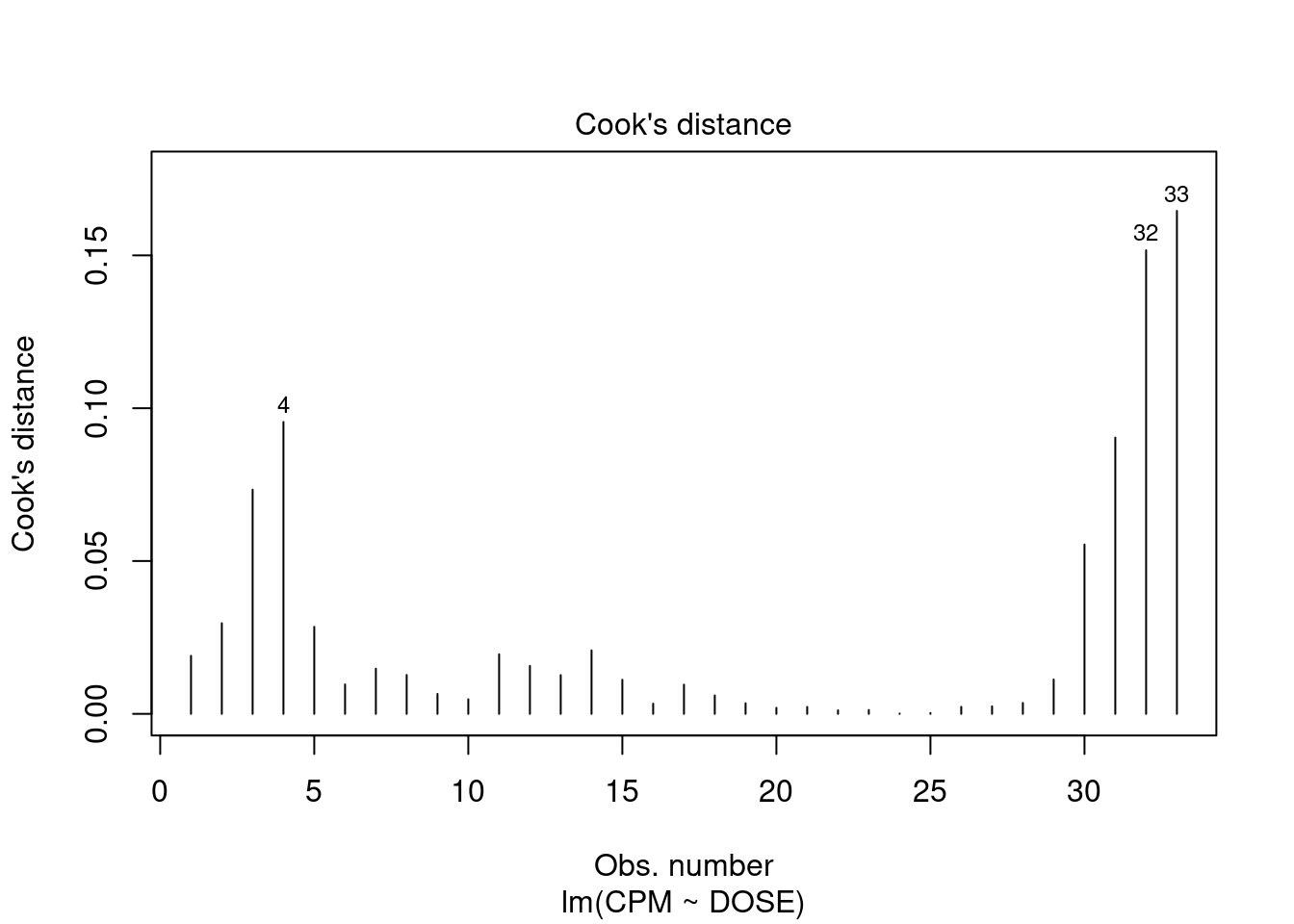
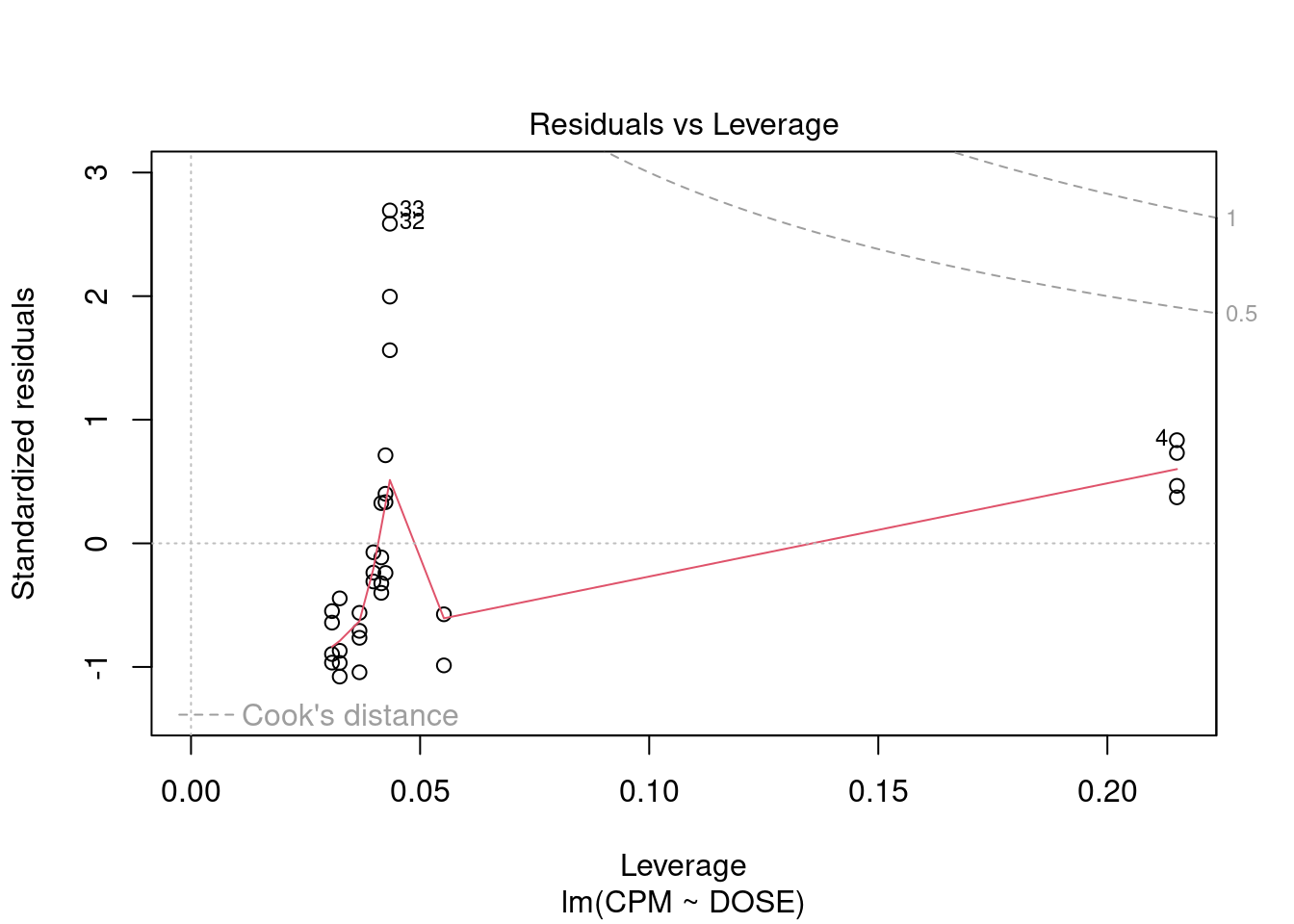
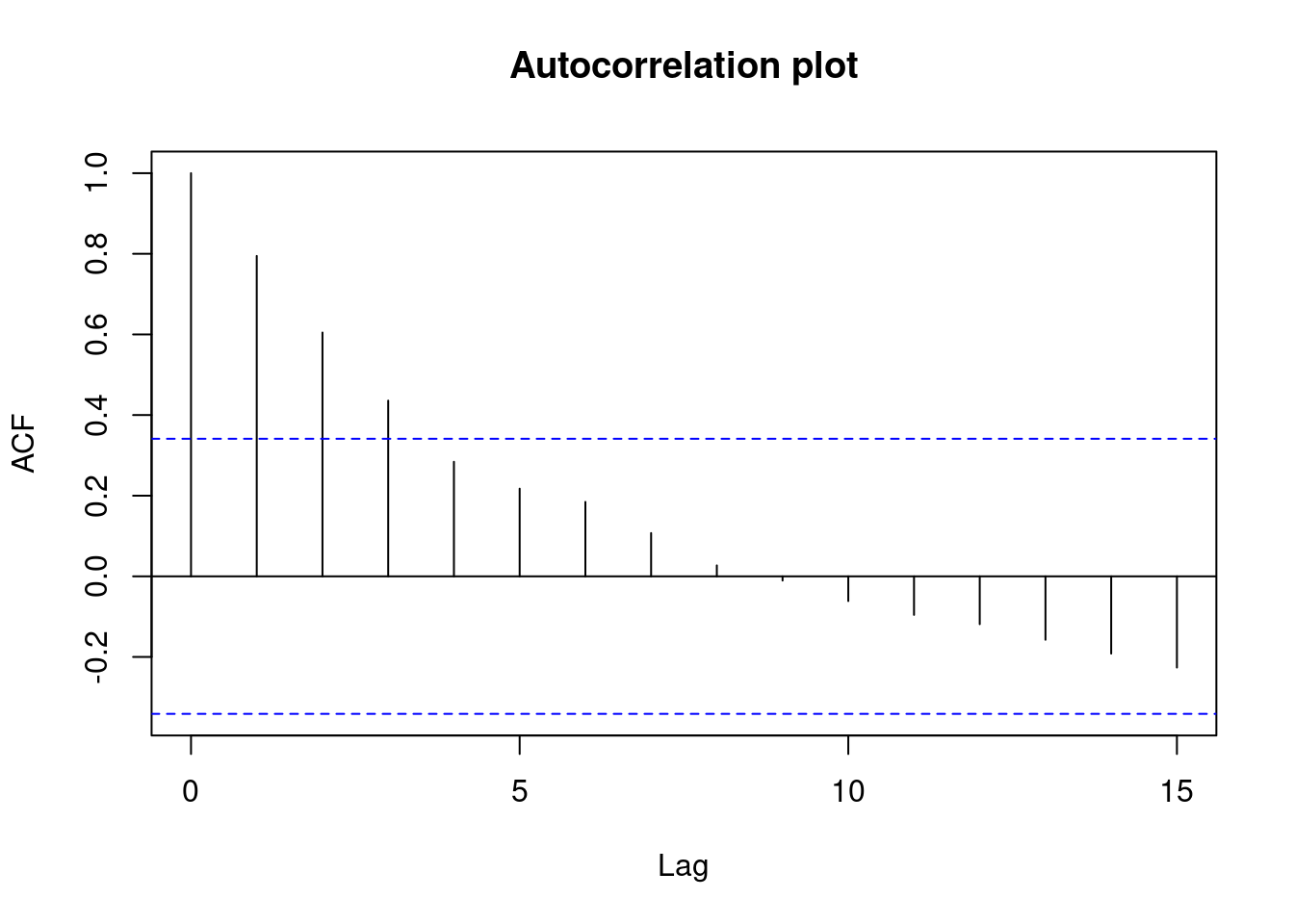
Perform regression diagnostics and test assumptions
Log transformation improves compliance with linear model assumptions. Some serial correlation remains, which is evident in plots of residuals vs fitted values and in the significant result of the Durbin-Watson D test, but it is within acceptable limits of the global validation of linear model assumptions.
##
## Call:
## lm(formula = log(CPM) ~ DOSE)
##
## Residuals:
## Min 1Q Median 3Q Max
## -0.57193 -0.11370 -0.02660 0.08659 0.46827
##
## Coefficients:
## Estimate Std. Error t value Pr(>|t|)
## (Intercept) 10.66086 0.04420 241.20 <2e-16 ***
## DOSE -0.05144 0.00231 -22.27 <2e-16 ***
## ---
## Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
##
## Residual standard error: 0.2122 on 31 degrees of freedom
## Multiple R-squared: 0.9412, Adjusted R-squared: 0.9393
## F-statistic: 495.8 on 1 and 31 DF, p-value: < 2.2e-16
##
##
## ASSESSMENT OF THE LINEAR MODEL ASSUMPTIONS
## USING THE GLOBAL TEST ON 4 DEGREES-OF-FREEDOM:
## Level of Significance = 0.05
##
## Call:
## gvlma(x = lmlog)
##
## Value p-value Decision
## Global Stat 3.952318 0.41250 Assumptions acceptable.
## Skewness 0.056280 0.81248 Assumptions acceptable.
## Kurtosis 0.676082 0.41094 Assumptions acceptable.
## Link Function 0.004124 0.94880 Assumptions acceptable.
## Heteroscedasticity 3.215833 0.07293 Assumptions acceptable.##
## Shapiro-Wilk normality test
##
## data: rstandard(lmlog)
## W = 0.94082, p-value = 0.07176## Warning in cor.test.default(fitted(lmlog), abs(rstandard(lmlog)), method =
## "spearman"): Cannot compute exact p-value with ties##
## Spearman's rank correlation rho
##
## data: fitted(lmlog) and abs(rstandard(lmlog))
## S = 6684, p-value = 0.5168
## alternative hypothesis: true rho is not equal to 0
## sample estimates:
## rho
## -0.1169866## lag Autocorrelation D-W Statistic p-value
## 1 0.4659162 0.7381887 0
## Alternative hypothesis: rho != 0## rstudent unadjusted p-value Bonferroni p
## 1 -3.574629 0.0012106 0.03995Simple linear regression jfs data
Feed intake vs total cholesterol data from Smith BW J Food Sci 2015 (doi: 10.1111/1750-3841.12968).
Import dataset
feedtc <- read.table('data/lm_jfs2015_feed_vs_tc.csv',
header = TRUE,
sep = ',')
attach(feedtc)
names(feedtc)## [1] "id" "diet" "tc" "feed"Perform regression diagnostics and test assumptions
The dataset contains some potential outliers (M1356 and M1376), and a violation of the link function according to gvlma.
- Outliers
- The major outlier is M1376, which returns a Cook’s D value of 0.43. This may or may not be an outlier, depending on the definition. It is just under the critical F value (F0.50(1,40)=0.463).
- The Bonferroni outlier test does not identify any outliers either.
- However, according to R in Action 8.4.3, Cook’s D values greater than 4/(n-k-1), where n is the sample size and k is the number of predictor variables, indicate influential observations (though he usually uses a cutoff of 1). M1376 would be considered an outlier by this definition.
- Link function
- The link function violation is difficult to address. The dataset conforms to a normal distribution as evidenced by the normal probability plot and Shapiro-Wilk W test, so it would not seem like a concern, but it is enough to violate gvlma. However, this doesn’t seem like a situation where it would be neccessary to switch to another model (such as a generalized linear mixed model) and specify a different link function.
- The Spearman rank order correlation between absolute values of residuals and predicted values returns a significant result, but heteroscedasticity is within acceptable limits according to gvlma and by visual inspection of plots of residuals vs. fitted values and autocorrelation.
##
## Call:
## lm(formula = feedtc$tc ~ feedtc$feed)
##
## Residuals:
## Min 1Q Median 3Q Max
## -465.42 -208.18 11.74 168.20 449.91
##
## Coefficients:
## Estimate Std. Error t value Pr(>|t|)
## (Intercept) -470.33 232.65 -2.022 0.0489 *
## feedtc$feed 77.39 14.94 5.179 4.58e-06 ***
## ---
## Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
##
## Residual standard error: 239.6 on 47 degrees of freedom
## Multiple R-squared: 0.3633, Adjusted R-squared: 0.3498
## F-statistic: 26.82 on 1 and 47 DF, p-value: 4.578e-06
##
##
## ASSESSMENT OF THE LINEAR MODEL ASSUMPTIONS
## USING THE GLOBAL TEST ON 4 DEGREES-OF-FREEDOM:
## Level of Significance = 0.05
##
## Call:
## gvlma(x = feedtclm1)
##
## Value p-value Decision
## Global Stat 15.17502 0.0043516 Assumptions NOT satisfied!
## Skewness 0.03692 0.8476301 Assumptions acceptable.
## Kurtosis 1.62599 0.2022584 Assumptions acceptable.
## Link Function 13.31102 0.0002639 Assumptions NOT satisfied!
## Heteroscedasticity 0.20109 0.6538402 Assumptions acceptable.##
## Shapiro-Wilk normality test
##
## data: rstandard(feedtclm1)
## W = 0.97342, p-value = 0.3295## Warning in cor.test.default(fitted(feedtclm1), abs(rstandard(feedtclm1)), :
## Cannot compute exact p-value with ties##
## Spearman's rank correlation rho
##
## data: fitted(feedtclm1) and abs(rstandard(feedtclm1))
## S = 25966, p-value = 0.02278
## alternative hypothesis: true rho is not equal to 0
## sample estimates:
## rho
## -0.3248125## lag Autocorrelation D-W Statistic p-value
## 1 0.4305706 1.065764 0
## Alternative hypothesis: rho != 0## No Studentized residuals with Bonferroni p < 0.05
## Largest |rstudent|:
## rstudent unadjusted p-value Bonferroni p
## 45 -2.208409 0.032238 NAGenerate plots
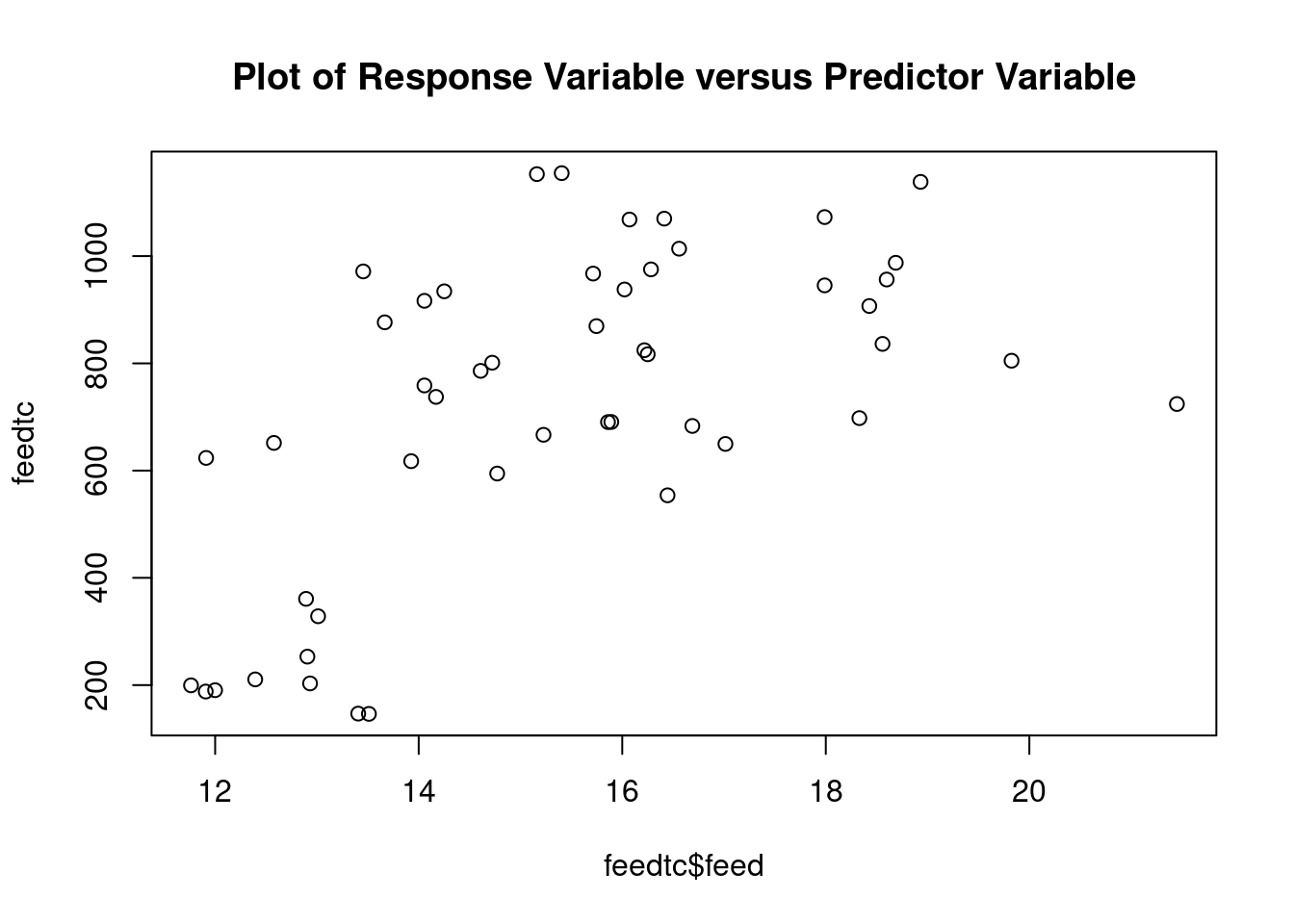
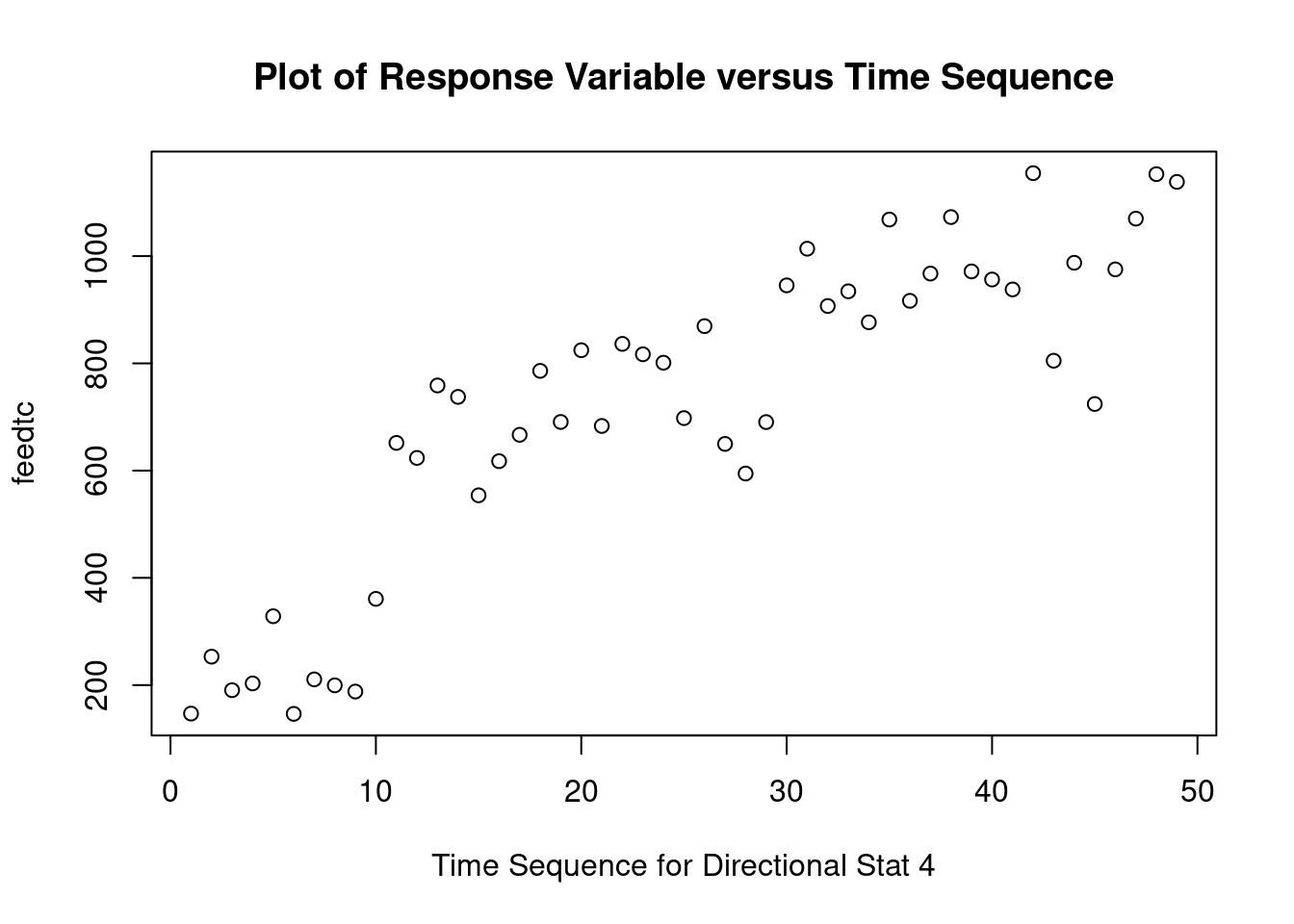
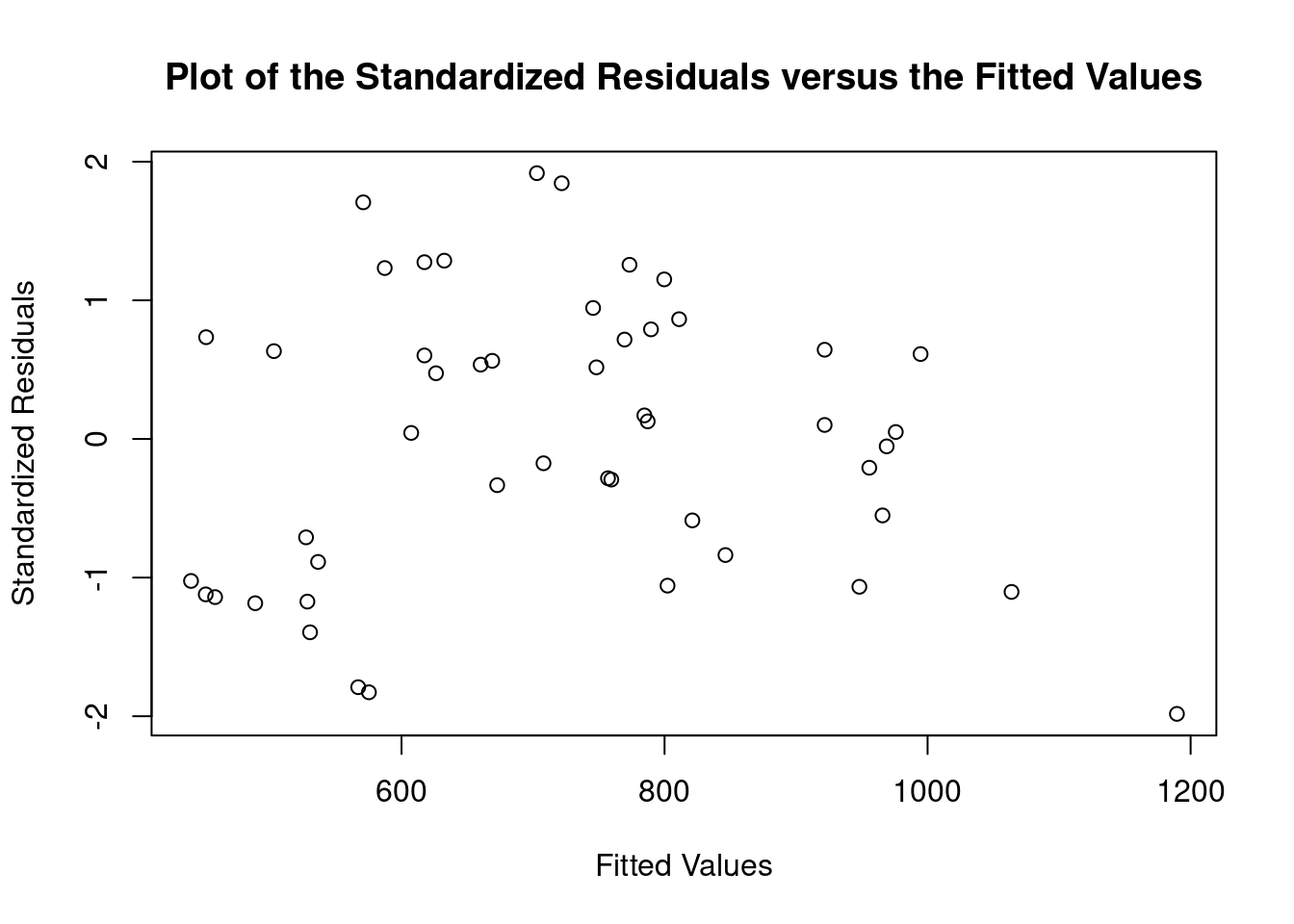
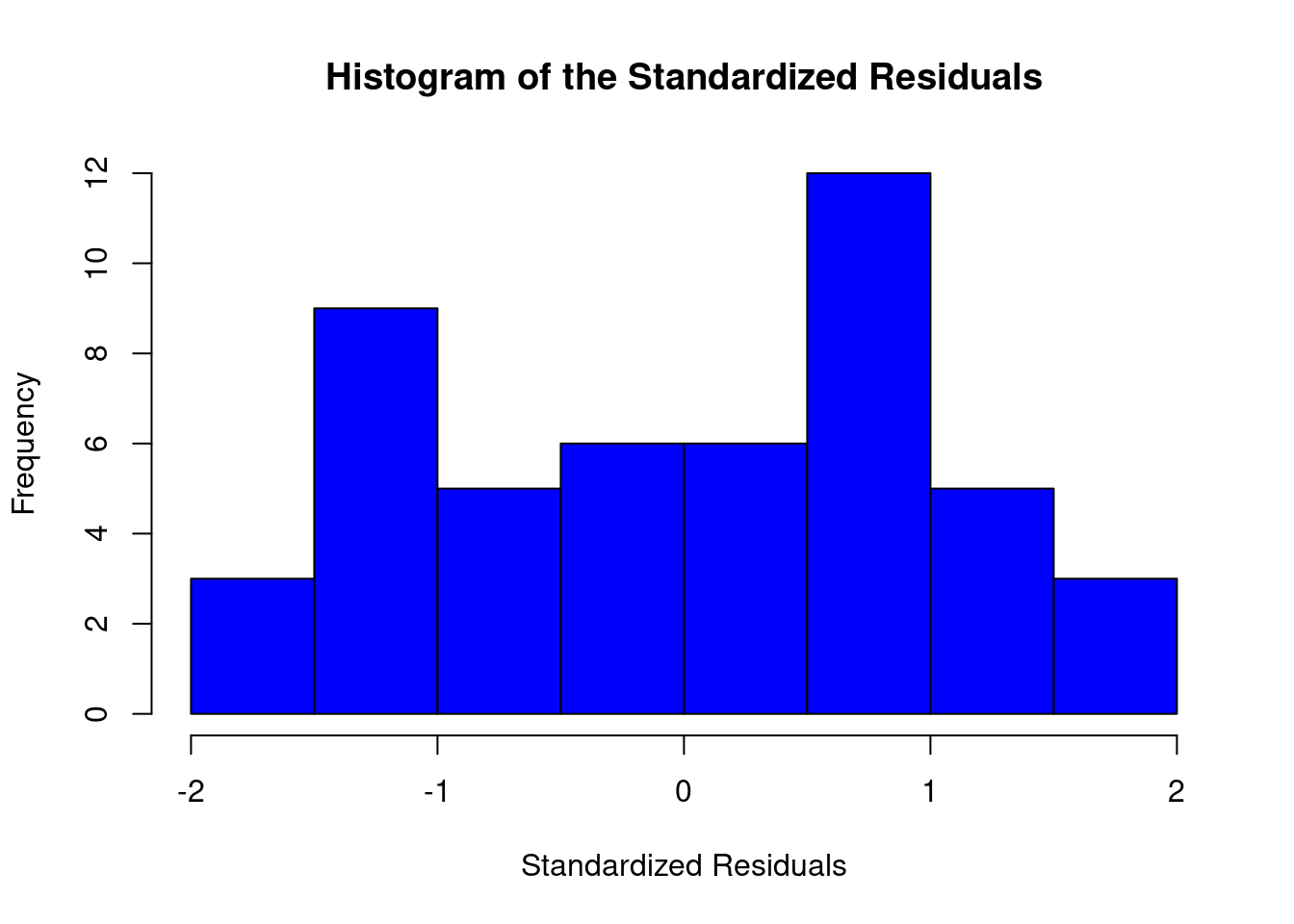
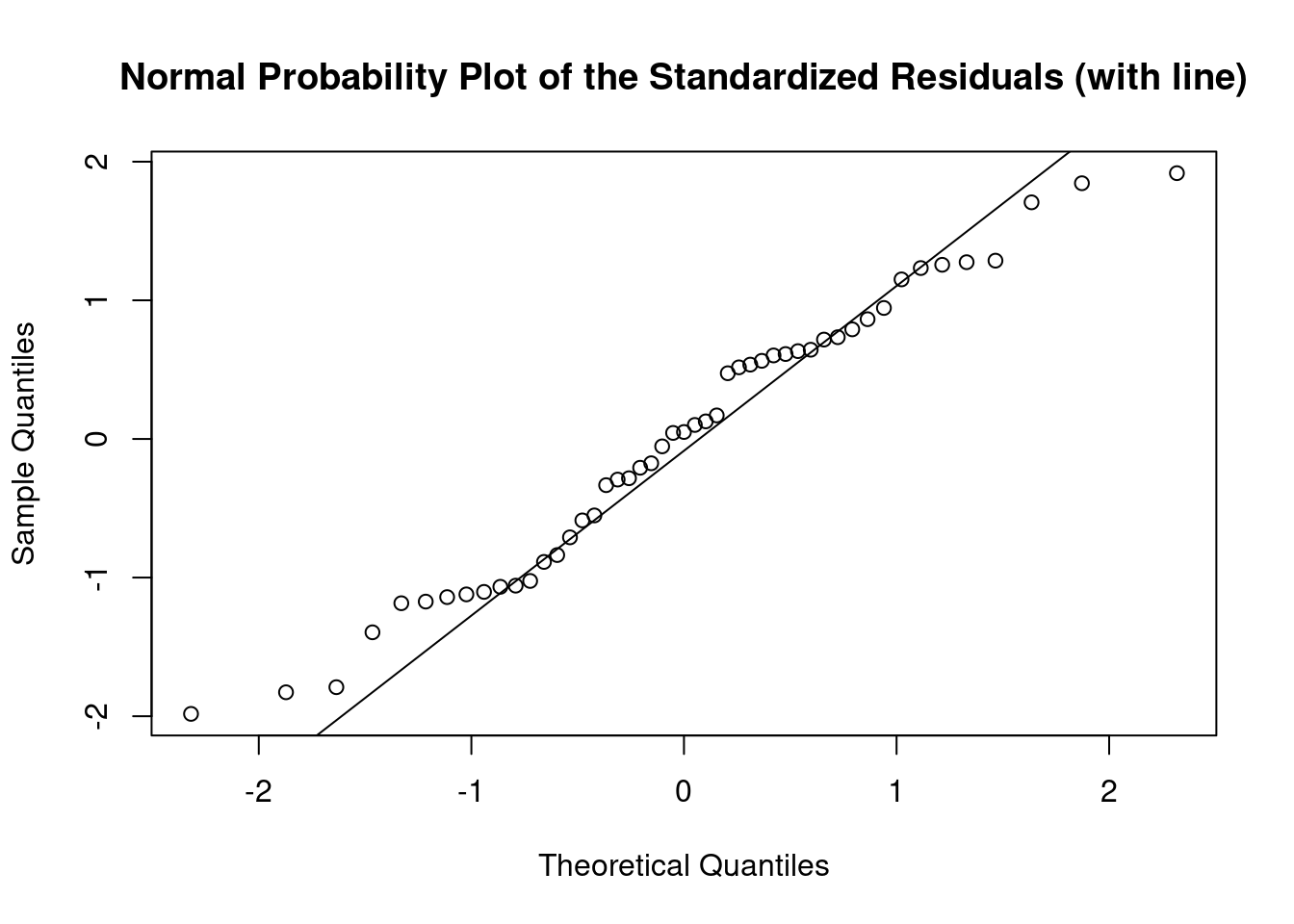
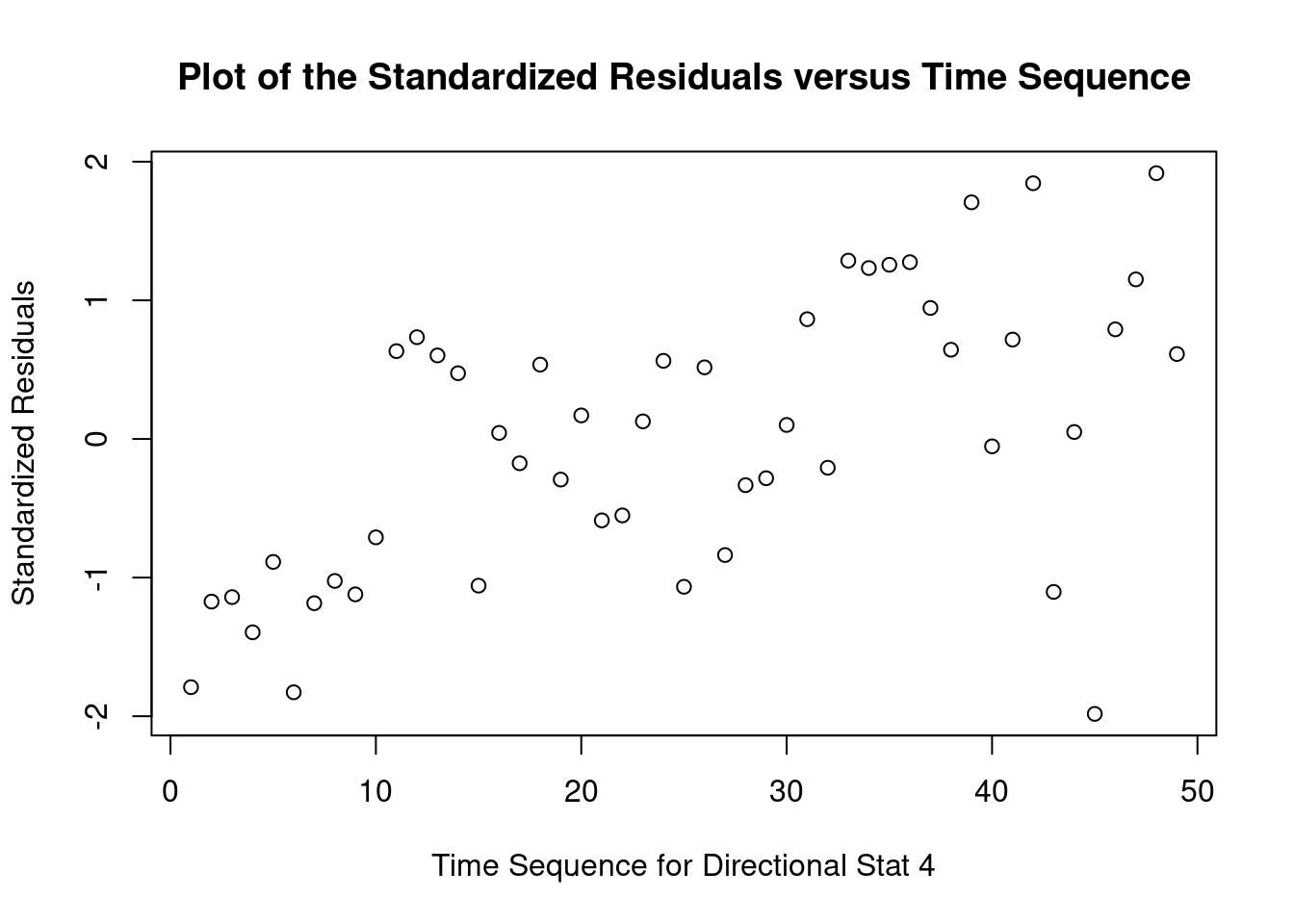
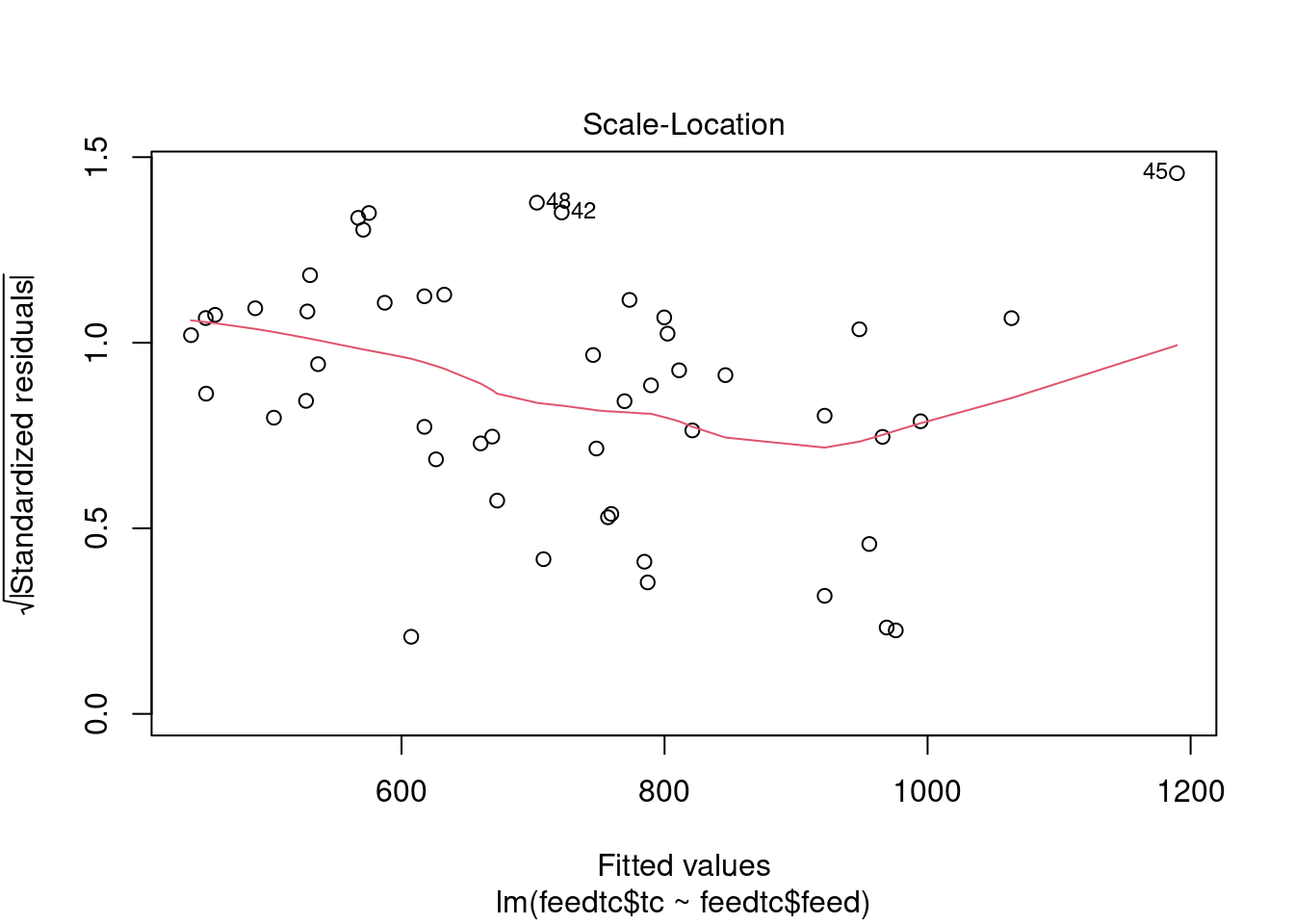
cookscritical <- 4 / (nrow(feedtc) - length(feedtclm1$coefficients) - 2)
plot(feedtclm1, which = 4, cook.levels = cookscritical)
abline(h = cookscritical, lty = 2, col = 'red')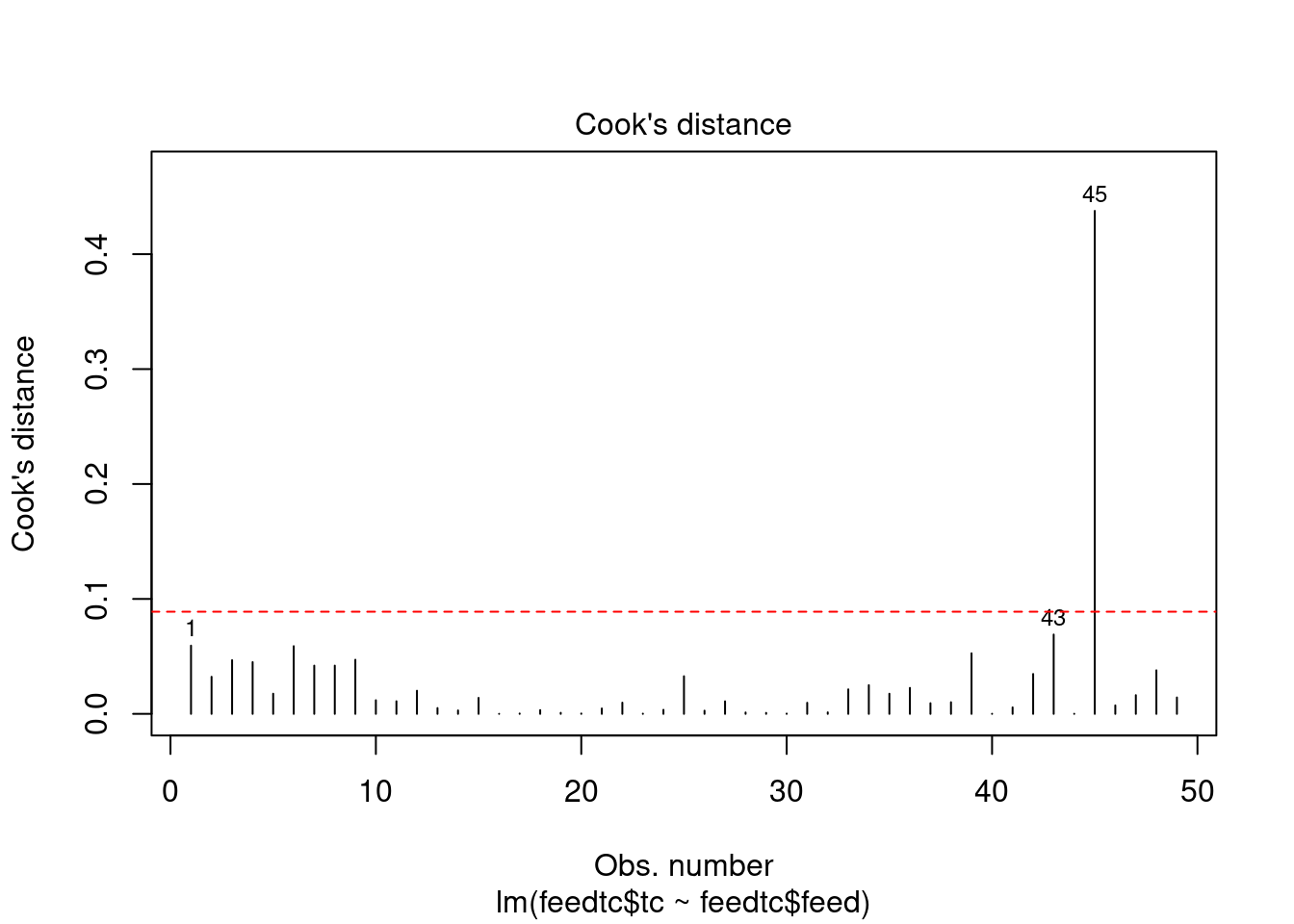
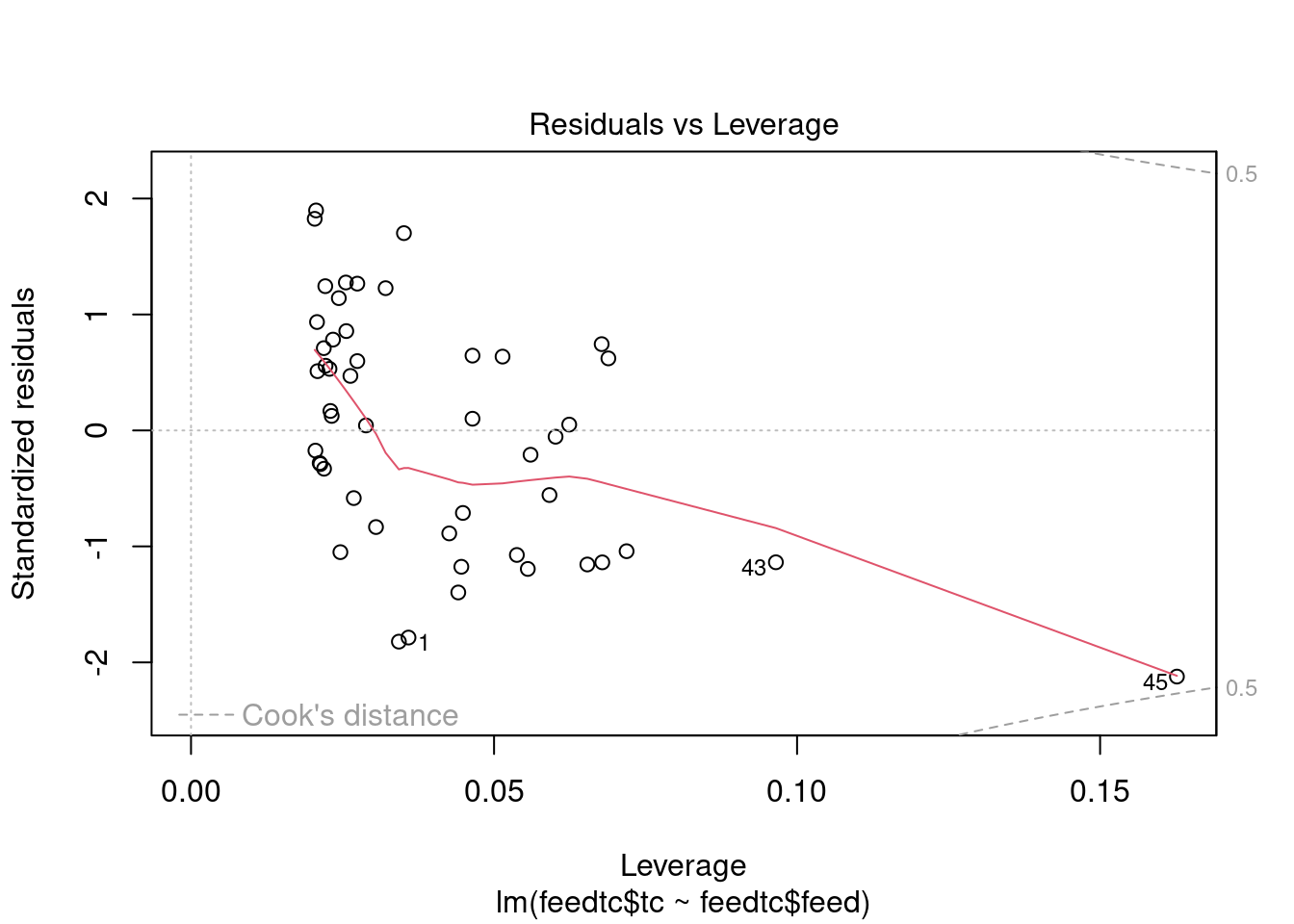
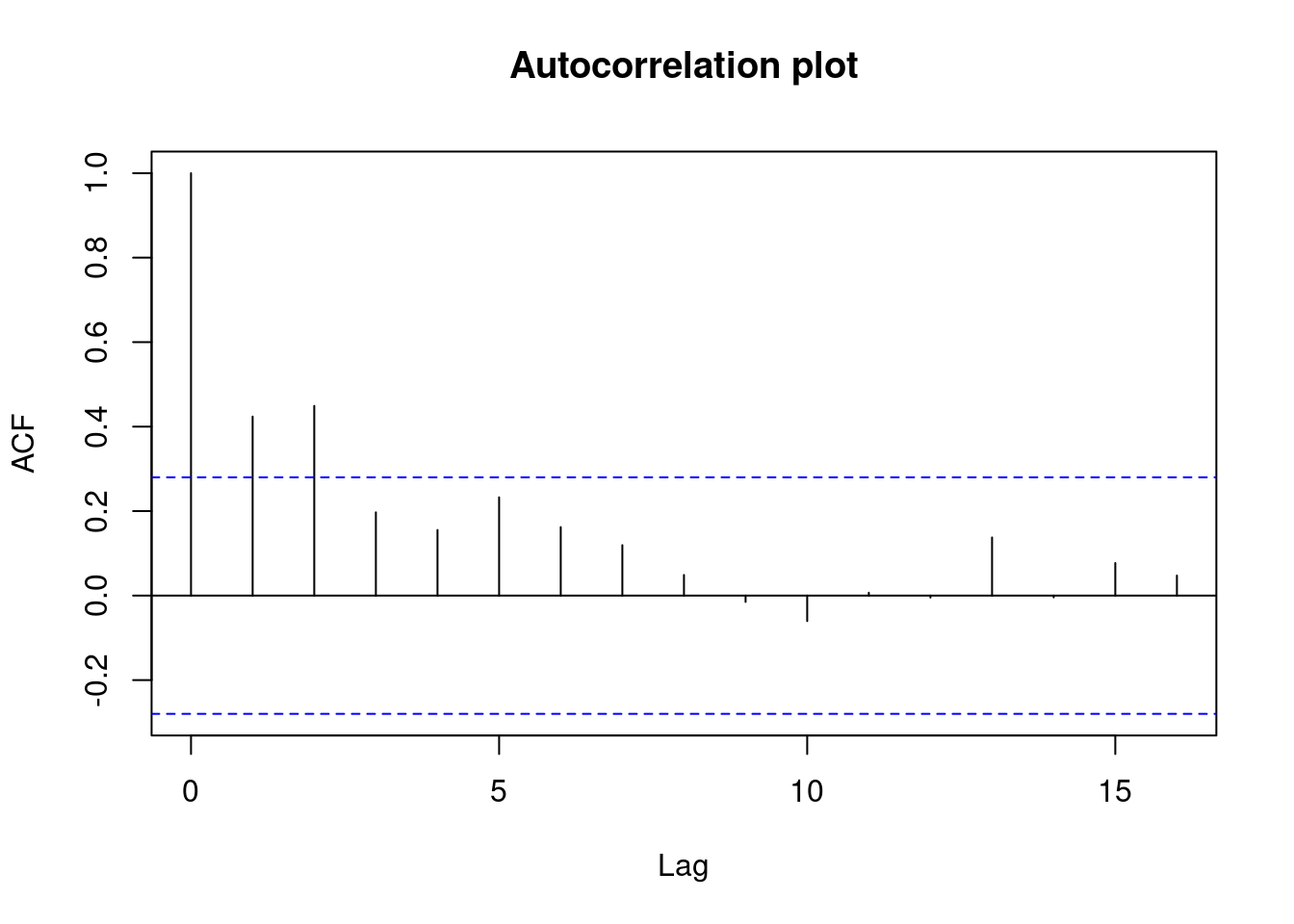
Calculate Spearman correlation
The Spearman rank order correlation between the independent and dependent variable is another type of correlation that is more widely valid, especially in situations where F test assumptions are violated.
## Warning in cor.test.default(feedtc$tc, feedtc$feed, method = "spearman"):
## Cannot compute exact p-value with ties##
## Spearman's rank correlation rho
##
## data: feedtc$tc and feedtc$feed
## S = 8028.4, p-value = 8.003e-06
## alternative hypothesis: true rho is not equal to 0
## sample estimates:
## rho
## 0.5903873Multiple linear regression and ANOVA example
This dataset from Doug Simpson at University of Illinois contains information on the number of calories in various beers, along with their percent alcohol and brewery.
PercentAlcohol is a quantitative continuous independent
variable and Brewery is a categorical independent variable.
Perform multiple linear regression in lm
## Analysis of Variance Table
##
## Response: beer$Calories
## Df Sum Sq Mean Sq F value Pr(>F)
## beer$PercentAlcohol 1 17914.2 17914.2 102.3698 7.9e-13 ***
## beer$Brewery 8 4424.9 553.1 3.1608 0.00682 **
## Residuals 42 7349.8 175.0
## ---
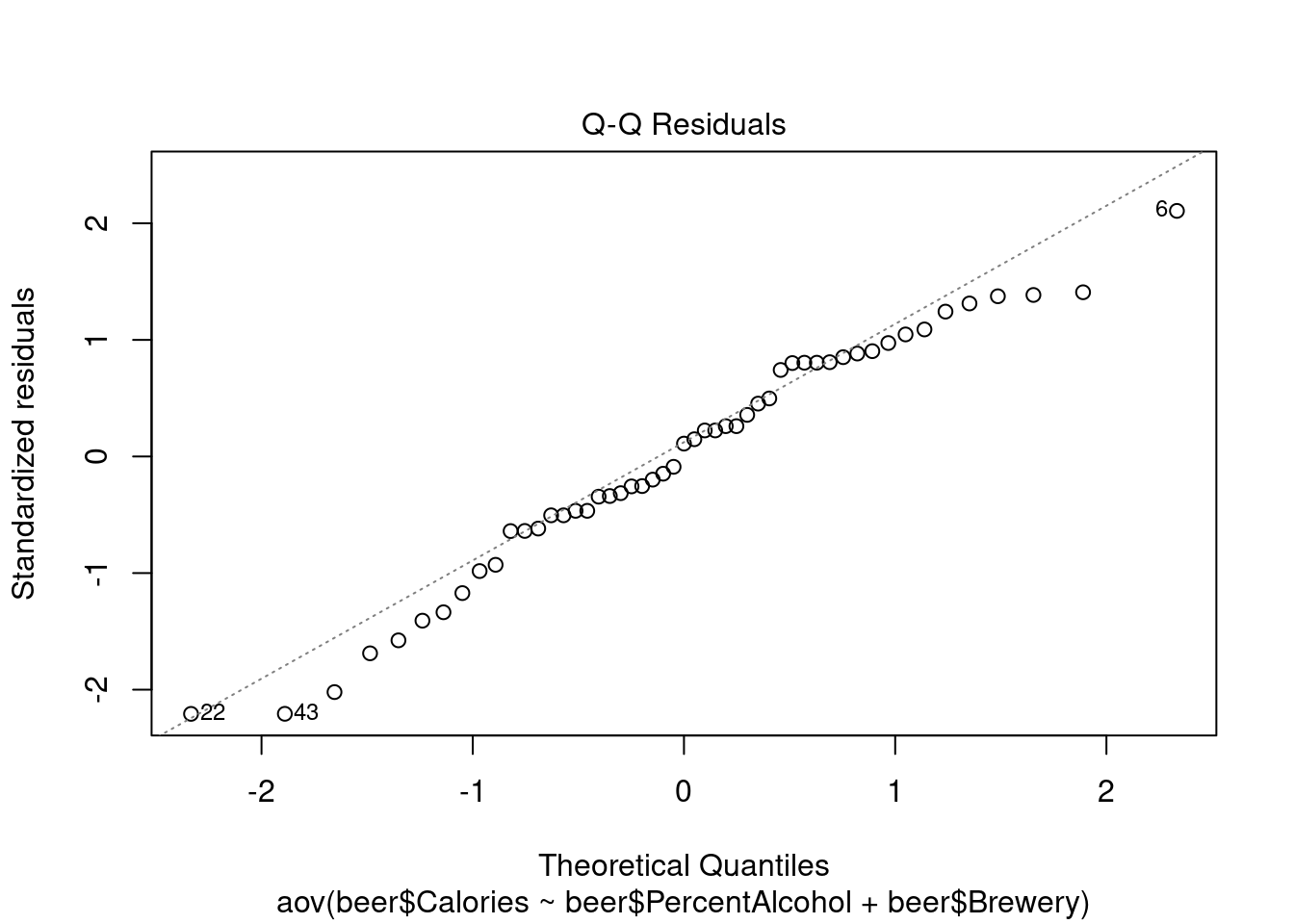
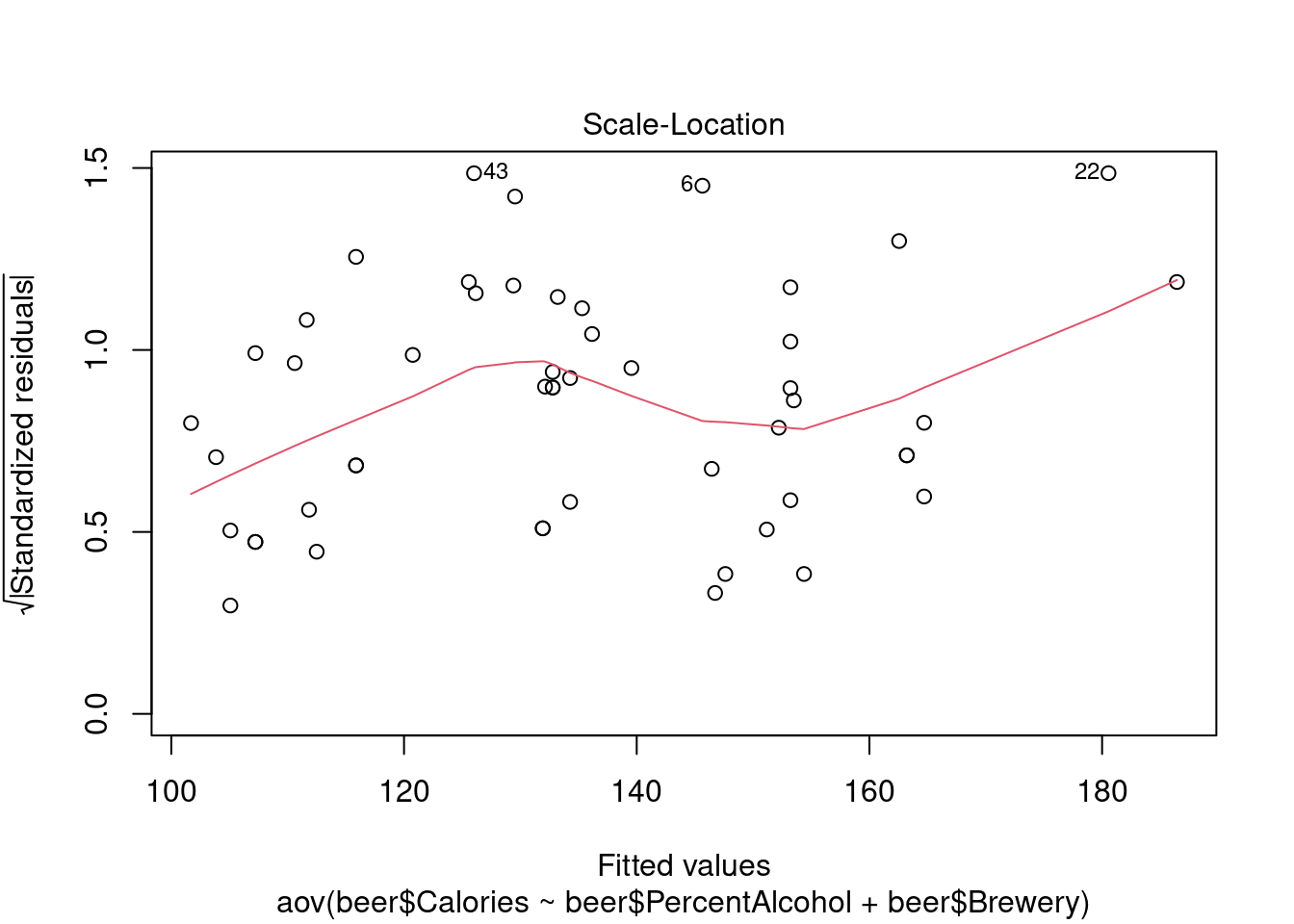
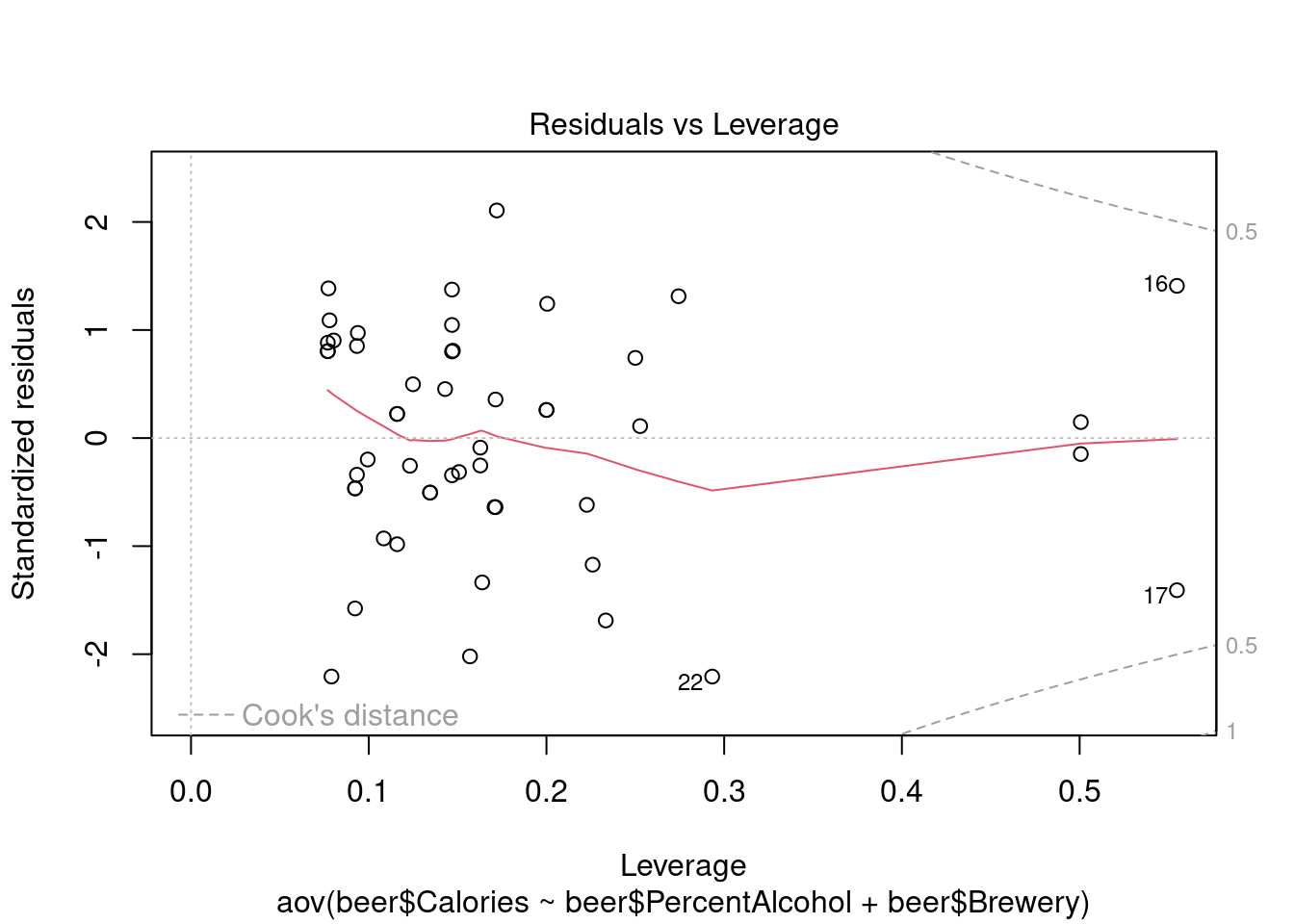
## Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1Perform regression diagnostics and test assumptions
##
## Call:
## lm(formula = beer$Calories ~ beer$PercentAlcohol + beer$Brewery)
##
## Residuals:
## Min 1Q Median 3Q Max
## -28.0179 -6.4610 0.6312 10.2197 25.3521
##
## Coefficients:
## Estimate Std. Error t value Pr(>|t|)
## (Intercept) -36.937 17.061 -2.165 0.03612 *
## beer$PercentAlcohol 33.812 3.439 9.831 1.86e-12 ***
## beer$BreweryAnchor 24.258 14.152 1.714 0.09389 .
## beer$BreweryAnheuser Busch 2.150 6.400 0.336 0.73858
## beer$BreweryD. G. Yuengling 20.088 7.825 2.567 0.01391 *
## beer$BreweryG. Heileman 17.115 10.722 1.596 0.11795
## beer$BreweryHigh Falls Brewing 17.996 8.382 2.147 0.03762 *
## beer$BreweryLeinenkugel 24.470 7.077 3.458 0.00126 **
## beer$BreweryMiller 10.801 6.206 1.740 0.08911 .
## beer$BreweryPabst 29.021 10.607 2.736 0.00908 **
## ---
## Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
##
## Residual standard error: 13.23 on 42 degrees of freedom
## Multiple R-squared: 0.7524, Adjusted R-squared: 0.6994
## F-statistic: 14.18 on 9 and 42 DF, p-value: 3.745e-10
##
##
## ASSESSMENT OF THE LINEAR MODEL ASSUMPTIONS
## USING THE GLOBAL TEST ON 4 DEGREES-OF-FREEDOM:
## Level of Significance = 0.05
##
## Call:
## gvlma(x = beerlm1)
##
## Value p-value Decision
## Global Stat 16.8557 0.0020618 Assumptions NOT satisfied!
## Skewness 1.0664 0.3017523 Assumptions acceptable.
## Kurtosis 0.3225 0.5700957 Assumptions acceptable.
## Link Function 14.4847 0.0001413 Assumptions NOT satisfied!
## Heteroscedasticity 0.9820 0.3216947 Assumptions acceptable.##
## Shapiro-Wilk normality test
##
## data: rstandard(beerlm1)
## W = 0.97308, p-value = 0.2957## Warning in cor.test.default(fitted(beerlm1), abs(rstandard(beerlm1)), method =
## "spearman"): Cannot compute exact p-value with ties##
## Spearman's rank correlation rho
##
## data: fitted(beerlm1) and abs(rstandard(beerlm1))
## S = 19184, p-value = 0.3561
## alternative hypothesis: true rho is not equal to 0
## sample estimates:
## rho
## 0.1319358## lag Autocorrelation D-W Statistic p-value
## 1 0.07781896 1.843075 0.192
## Alternative hypothesis: rho != 0## Warning in leveneTest.default(beer$Calories, beer$Brewery): beer$Brewery
## coerced to factor.## Levene's Test for Homogeneity of Variance (center = median)
## Df F value Pr(>F)
## group 8 0.8577 0.5587
## 43Generate plots
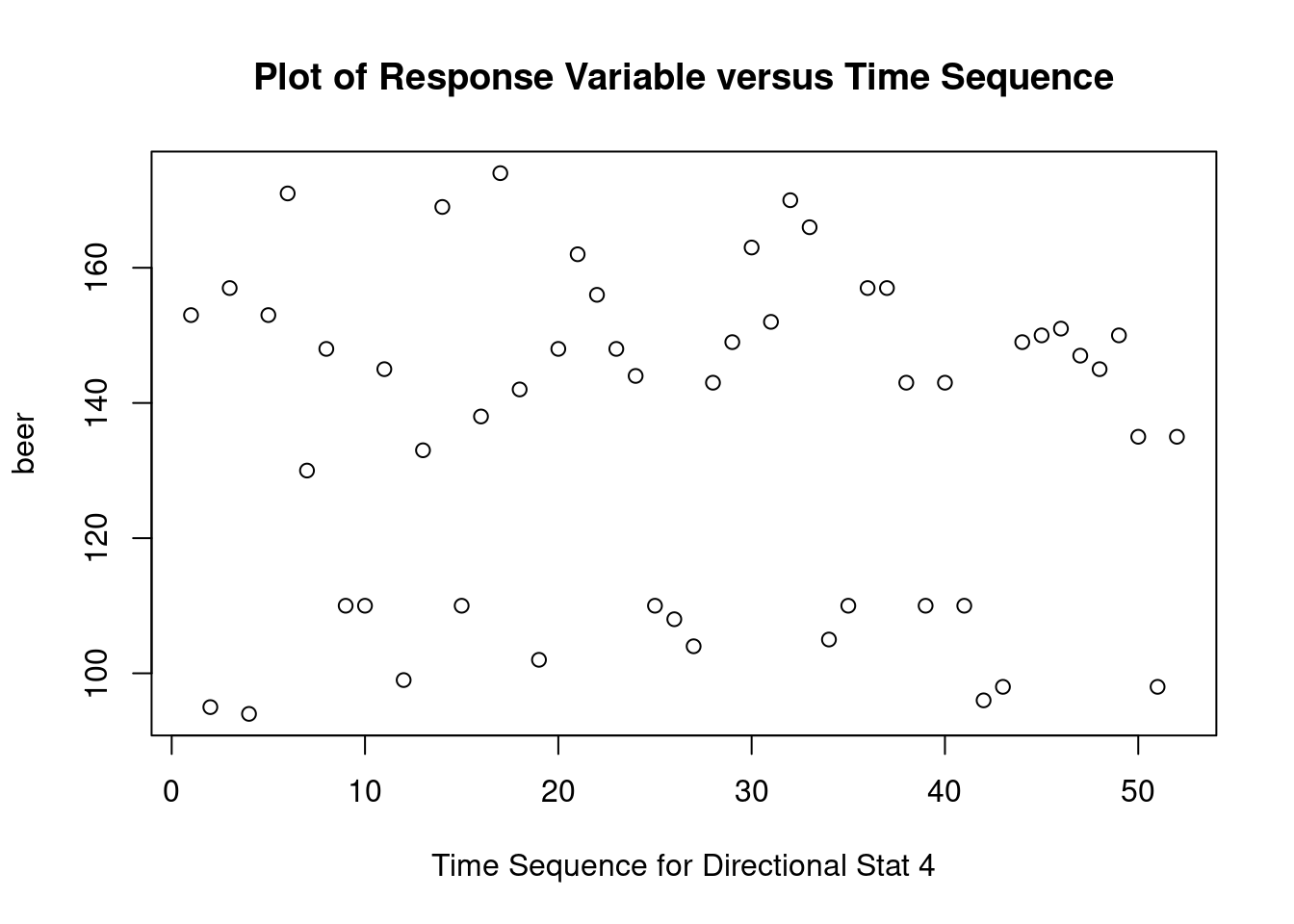
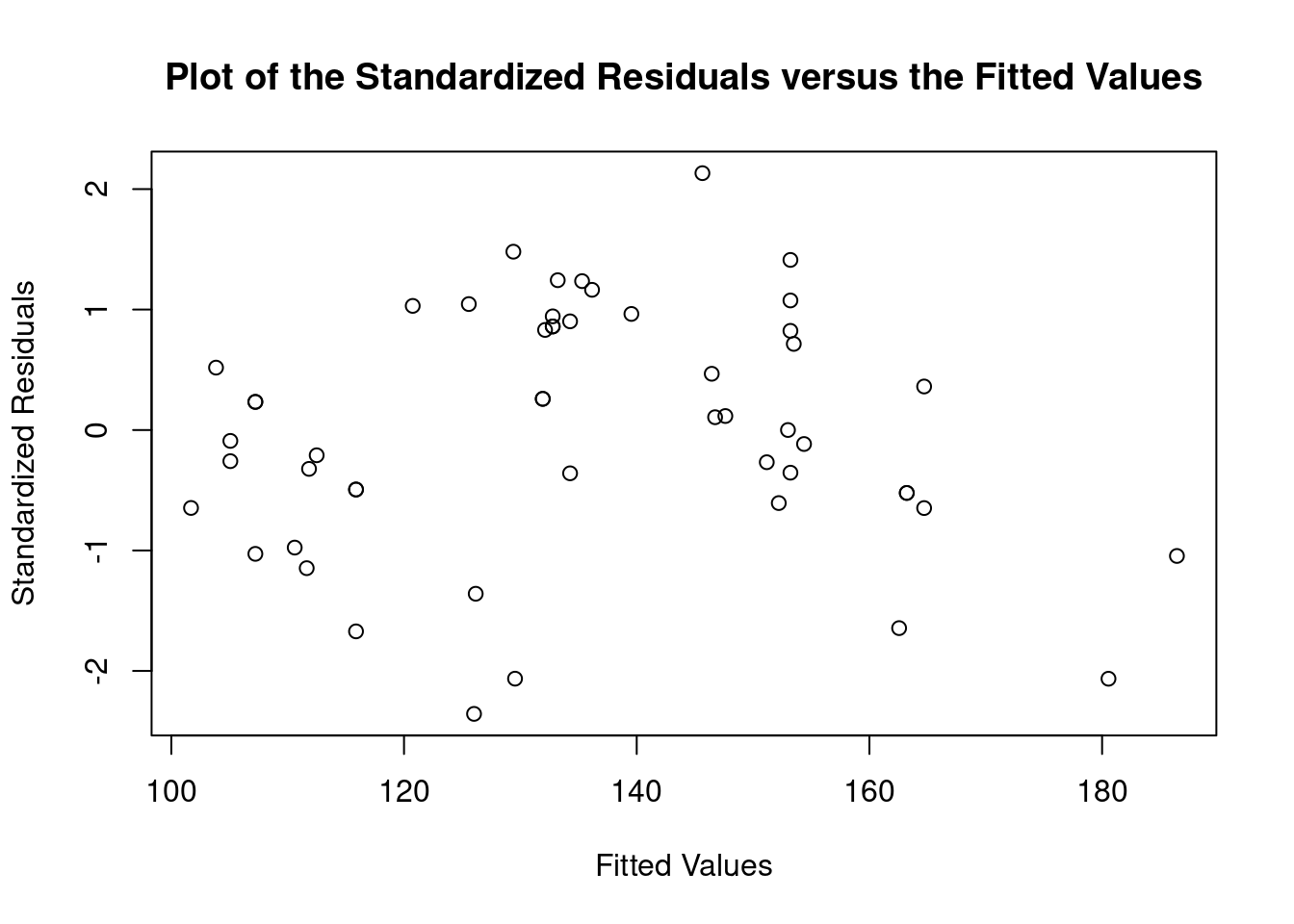
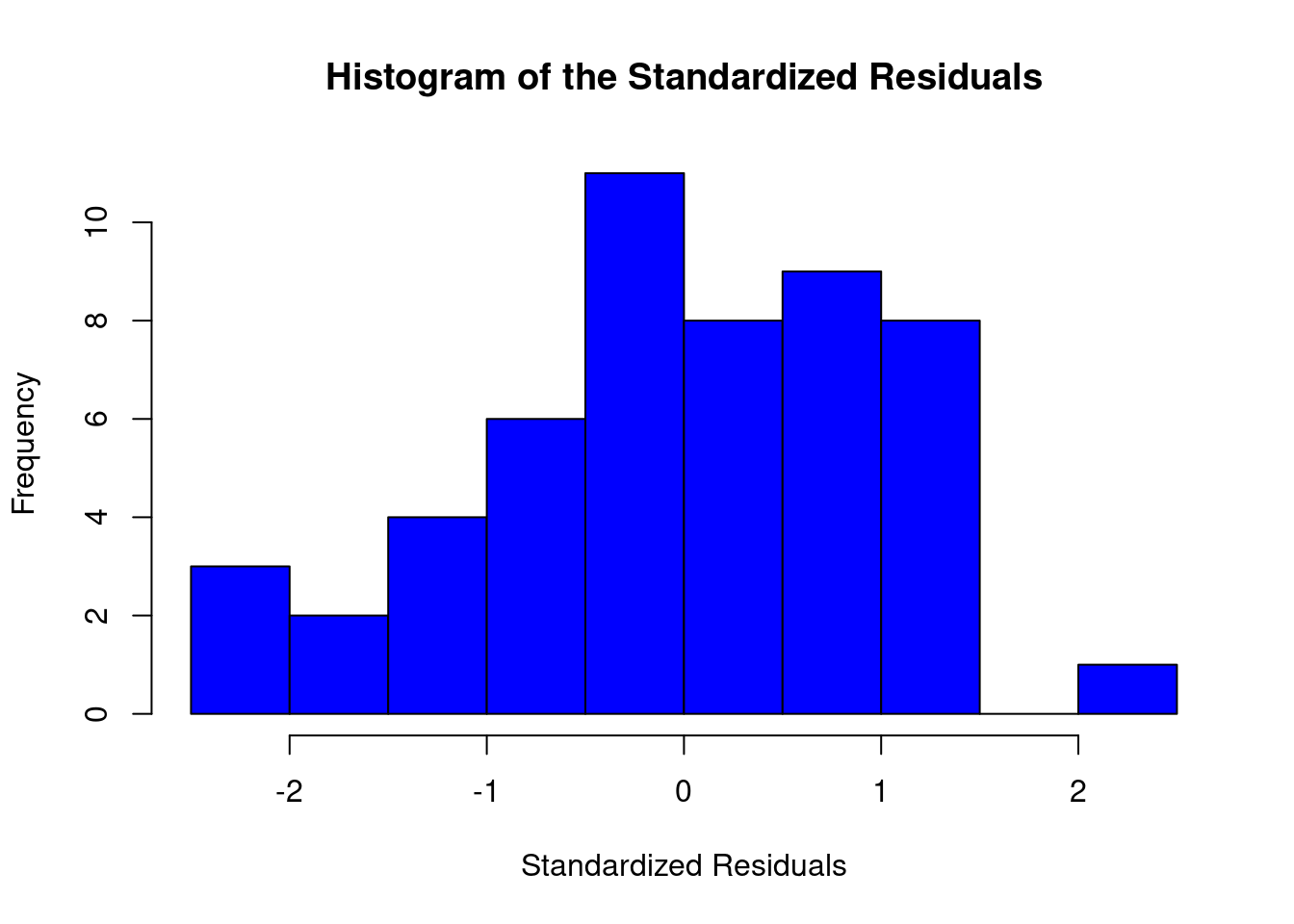
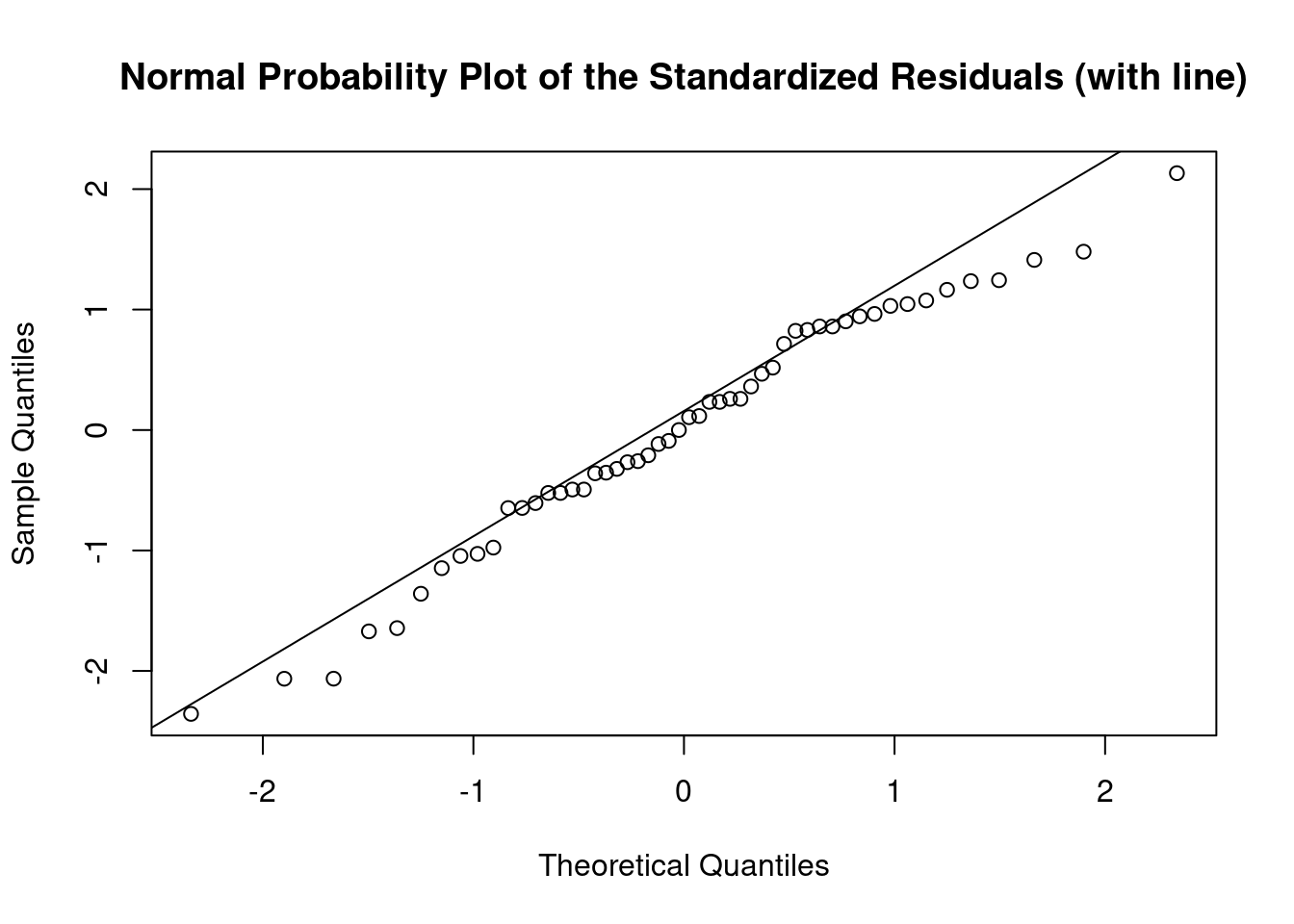
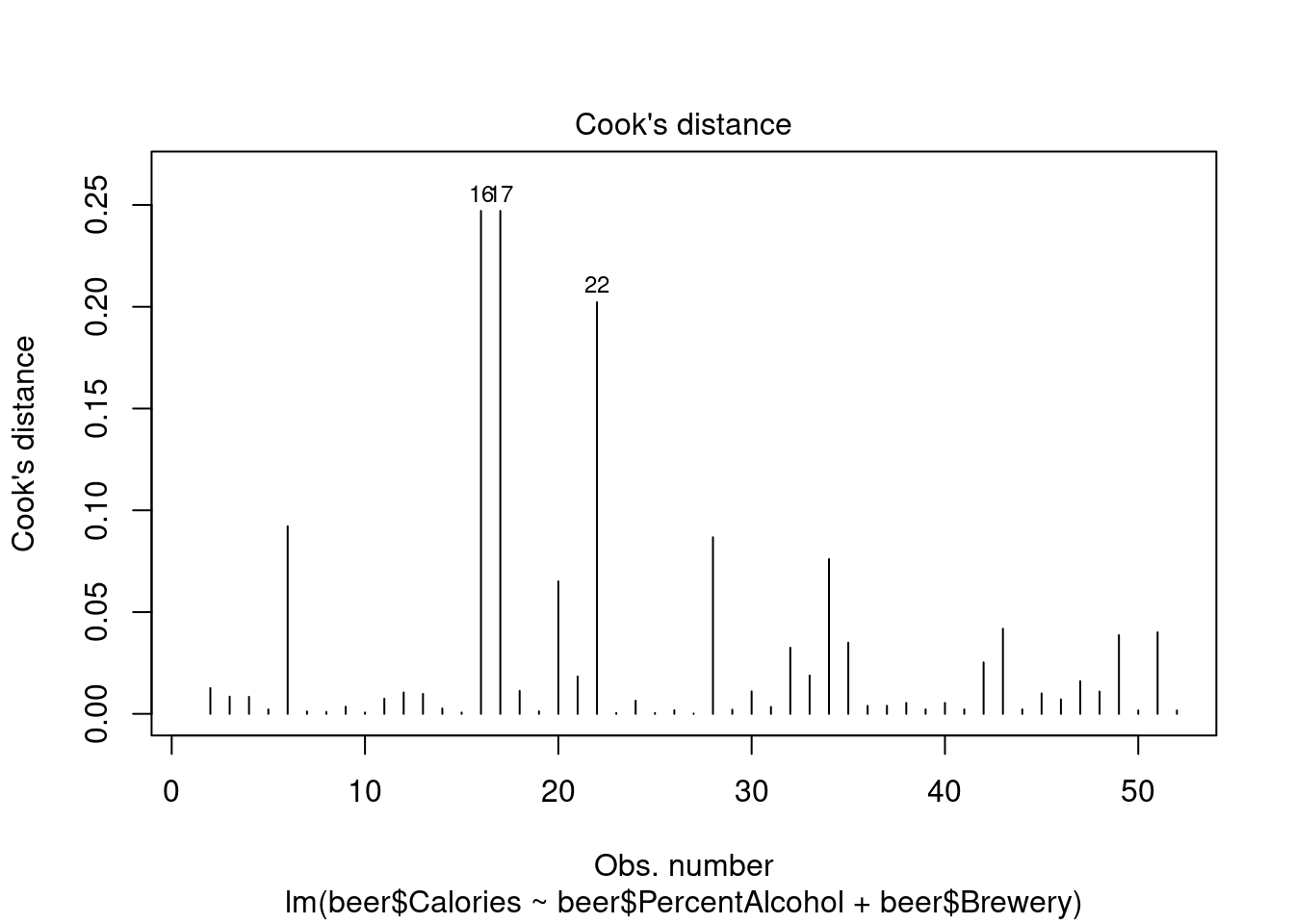
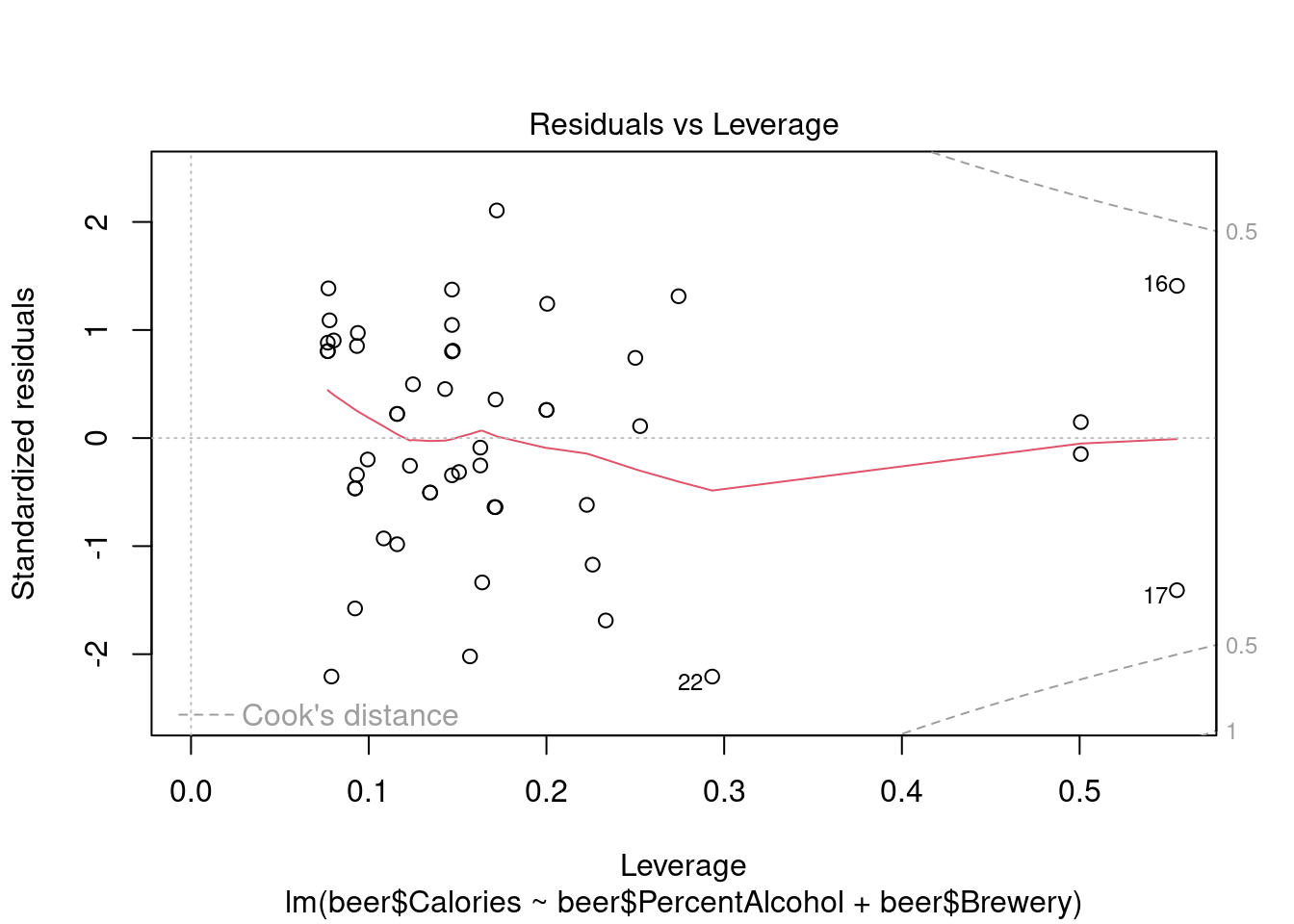
## Warning: not plotting observations with leverage one:
## 1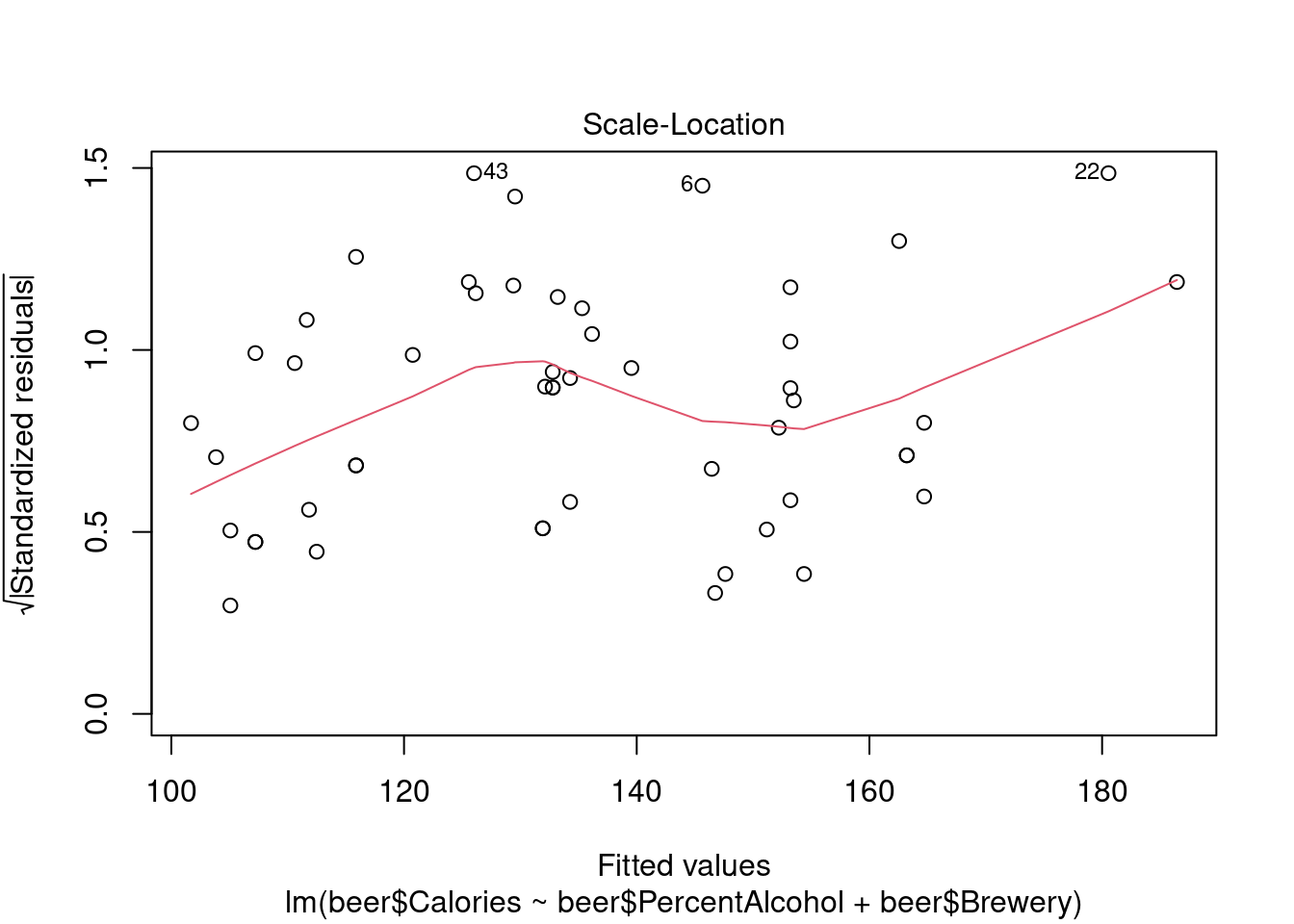
Perform ANOVA in aov for comparison
## Df Sum Sq Mean Sq F value Pr(>F)
## beer$PercentAlcohol 1 17914 17914 102.370 7.9e-13 ***
## beer$Brewery 8 4425 553 3.161 0.00682 **
## Residuals 42 7350 175
## ---
## Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1## Warning: not plotting observations with leverage one:
## 1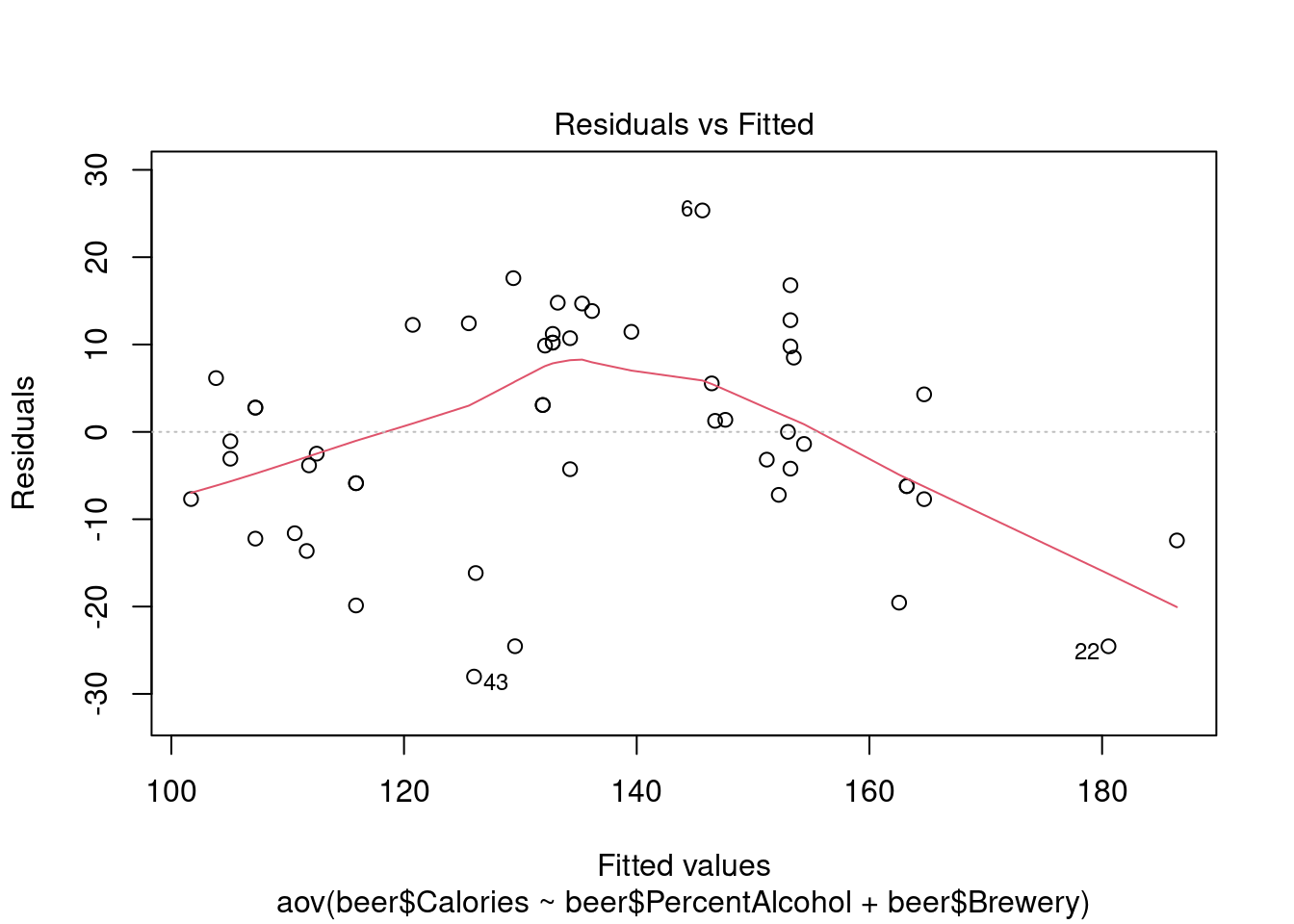
ANOVA
One-way ANOVA example
Gonadal adipose weights from Smith BW J Food Sci 2015 (doi: 10.1111/1750-3841.12968).
Import dataset
adipose_weights <-
read.table('data/anova_one_way_jfs2015_adipose.csv',
header = TRUE,
sep = ',')
attach(adipose_weights)
names(adipose_weights)## [1] "id" "diet" "adipose"Perform ANOVA in aov
# Convert independent variable to a factor for use later by glht()
adipose_weights$diet <- as.factor(adipose_weights$diet)
# Perform ANOVA
adiposeaov1 <- aov(adipose ~ diet, data = adipose_weights)
summary(adiposeaov1)## Df Sum Sq Mean Sq F value Pr(>F)
## diet 4 0.002509 0.0006271 9.949 9.35e-06 ***
## Residuals 42 0.002648 0.0000630
## ---
## Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
## 2 observations deleted due to missingnessPerform multiple comparisons
- The RColorBrewer package is loaded for additional colors, and a
palette is added to R. Use brackets after vector name for specific
colors when creating plots (for example:
col=set1[4:14]). - I chose the
Pastel1palette. “I’m a very vivacious man… Earth tones are so passe. I’m a pastel man.” Larry David, Curb Your Enthusiasm S01E05 “Interior Decorator”
## Tukey multiple comparisons of means
## 95% family-wise confidence level
##
## Fit: aov(formula = adipose ~ diet, data = adipose_weights)
##
## $diet
## diff lwr upr p adj
## WD-AIN 0.013272442 0.002278112 0.024266773 0.0109966
## WDSG-AIN 0.007630272 -0.003102252 0.018362795 0.2716464
## WDTP-AIN 0.018925192 0.008192669 0.029657715 0.0000924
## WDTPSG-AIN 0.020266814 0.009534291 0.030999337 0.0000292
## WDSG-WD -0.005642171 -0.016038167 0.004753826 0.5389261
## WDTP-WD 0.005652750 -0.004743247 0.016048746 0.5371109
## WDTPSG-WD 0.006994372 -0.003401625 0.017390368 0.3242717
## WDTP-WDSG 0.011294920 0.001176200 0.021413640 0.0219233
## WDTPSG-WDSG 0.012636543 0.002517823 0.022755262 0.0079286
## WDTPSG-WDTP 0.001341622 -0.008777098 0.011460342 0.9955000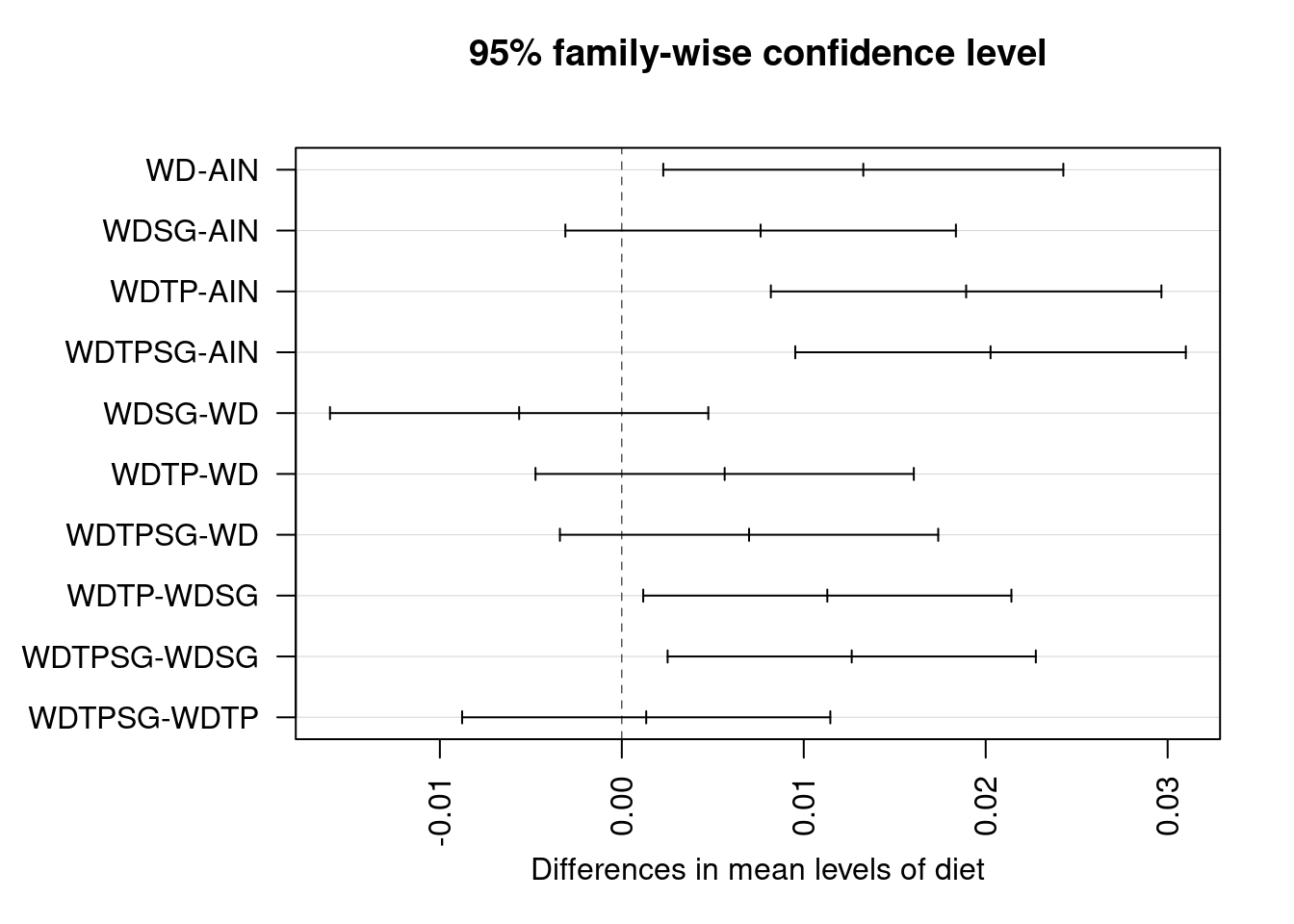
# More comprehensive method in multcomp package
par(las = 1, mar = c(5, 4, 6, 2))
library(multcomp)
library(RColorBrewer)
tuk <- glht(adiposeaov1, linfct = mcp(diet = 'Tukey'))
means <- aggregate(adipose ~ diet, adipose_weights, mean)
plot(cld(tuk, level = .05), col = brewer.pal(8, 'Pastel1'))
points(means, pch = 23)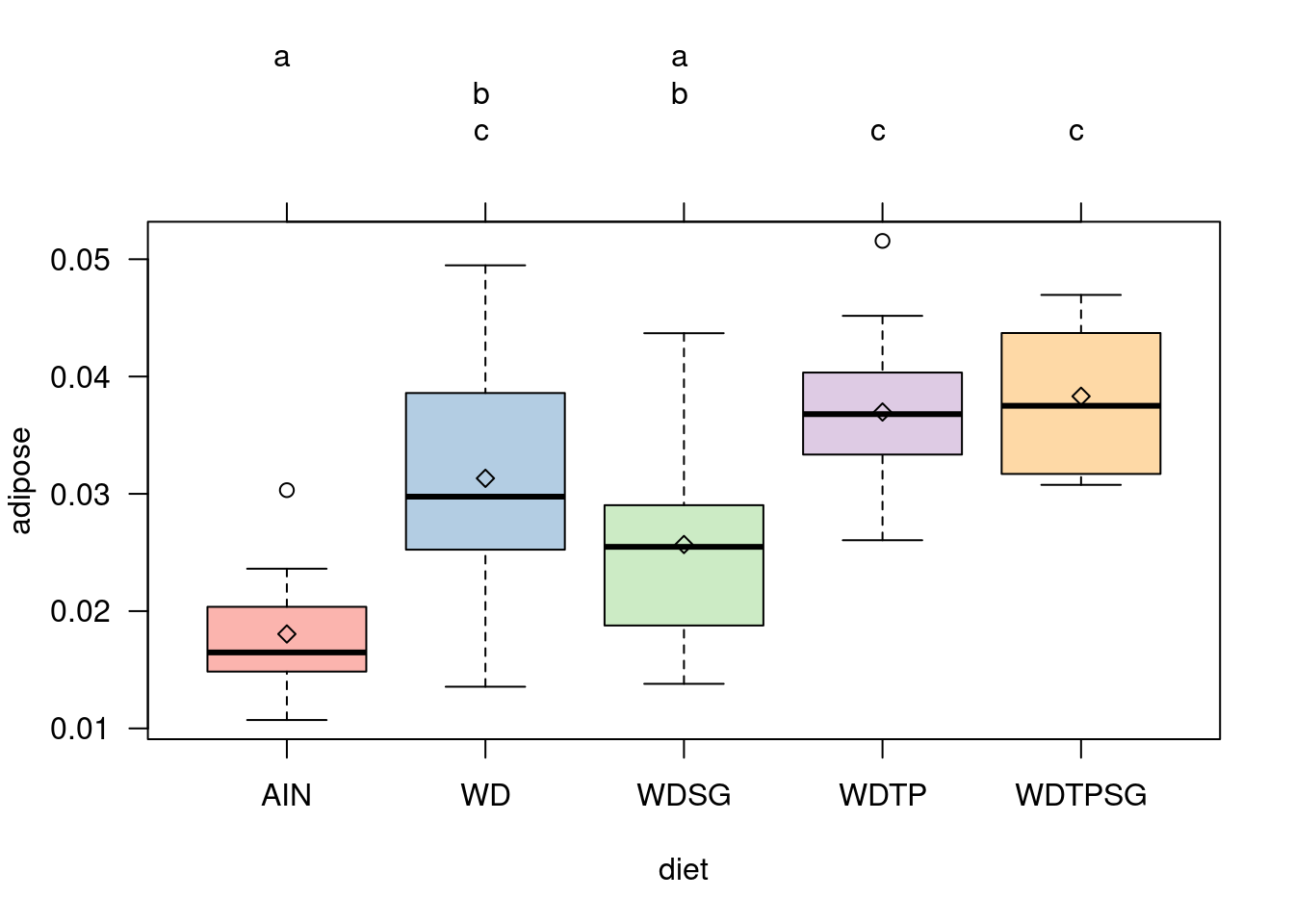
Test assumptions
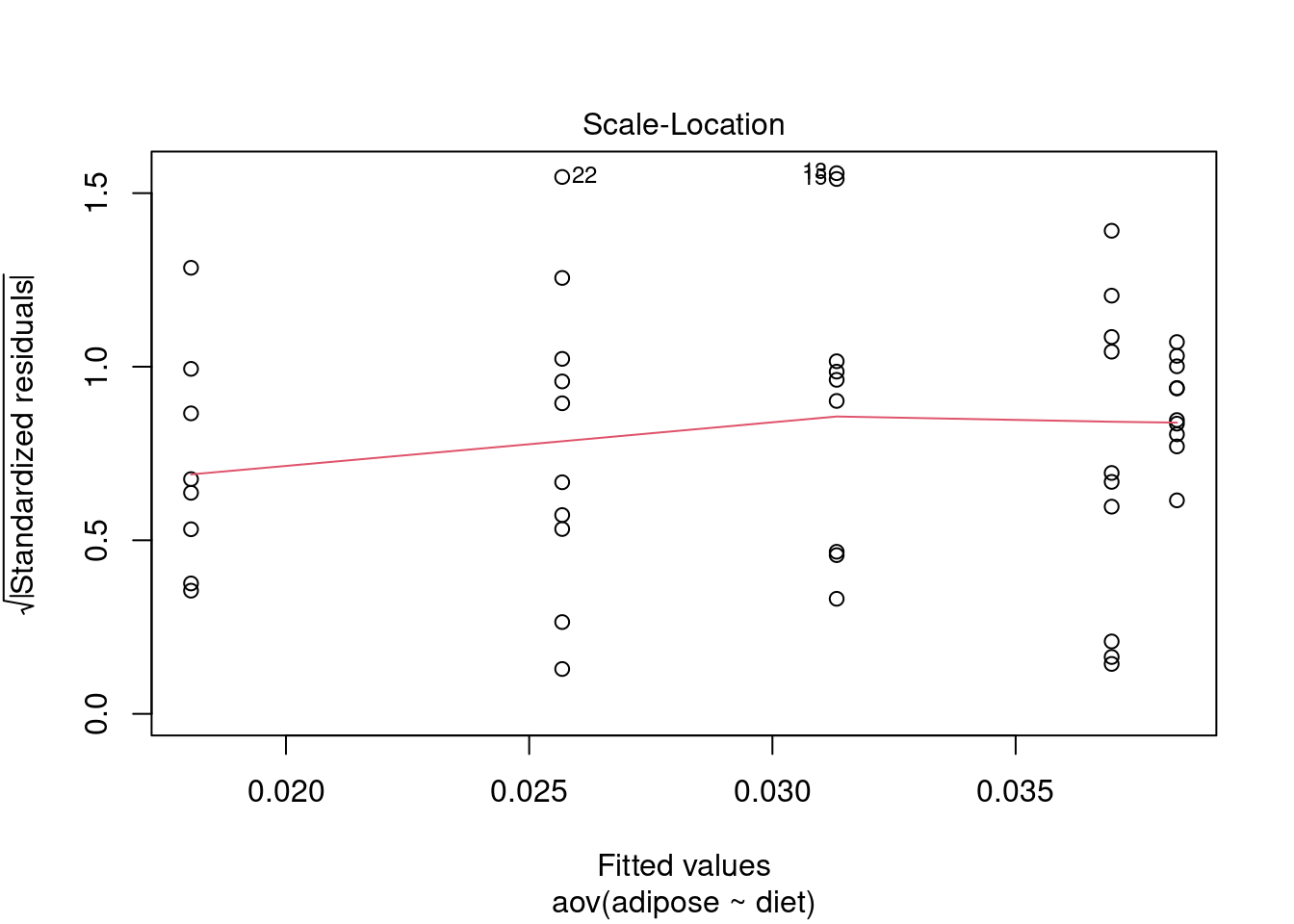
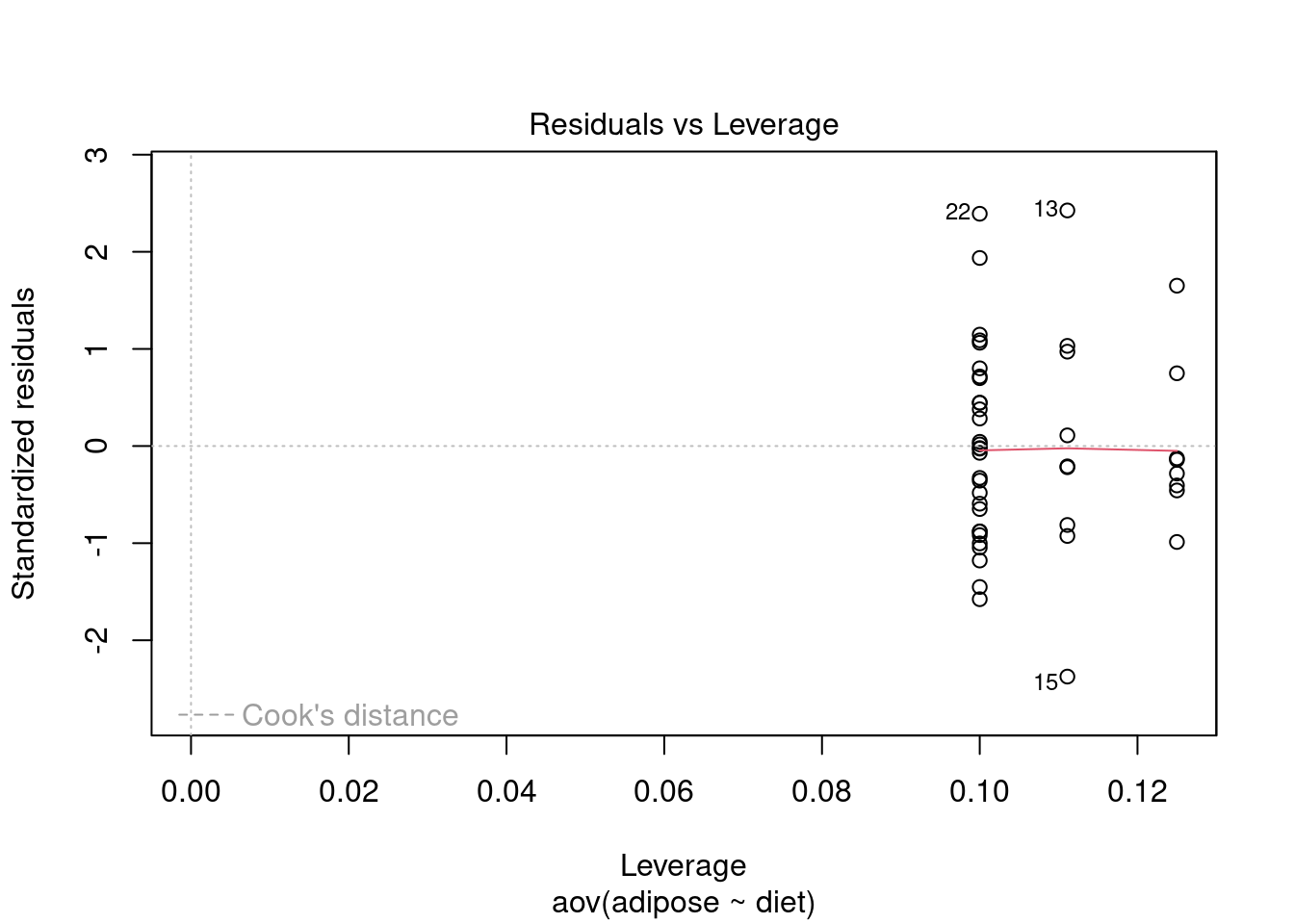
Normality
##
## Shapiro-Wilk normality test
##
## data: rstandard(adiposeaov1)
## W = 0.97688, p-value = 0.4704Perform ANOVA in lm for comparison
## Analysis of Variance Table
##
## Response: adipose_weights$adipose
## Df Sum Sq Mean Sq F value Pr(>F)
## adipose_weights$diet 4 0.0025086 0.00062714 9.9489 9.349e-06 ***
## Residuals 42 0.0026475 0.00006304
## ---
## Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1Perform regression diagnostics and test assumptions
##
## Call:
## lm(formula = adipose_weights$adipose ~ adipose_weights$diet)
##
## Residuals:
## Min 1Q Median 3Q Max
## -0.0177688 -0.0054835 -0.0009354 0.0053295 0.0181588
##
## Coefficients:
## Estimate Std. Error t value Pr(>|t|)
## (Intercept) 0.018048 0.002807 6.430 9.57e-08 ***
## adipose_weights$dietWD 0.013272 0.003858 3.440 0.00133 **
## adipose_weights$dietWDSG 0.007630 0.003766 2.026 0.04914 *
## adipose_weights$dietWDTP 0.018925 0.003766 5.025 9.81e-06 ***
## adipose_weights$dietWDTPSG 0.020267 0.003766 5.381 3.06e-06 ***
## ---
## Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
##
## Residual standard error: 0.00794 on 42 degrees of freedom
## (2 observations deleted due to missingness)
## Multiple R-squared: 0.4865, Adjusted R-squared: 0.4376
## F-statistic: 9.949 on 4 and 42 DF, p-value: 9.349e-06
##
##
## ASSESSMENT OF THE LINEAR MODEL ASSUMPTIONS
## USING THE GLOBAL TEST ON 4 DEGREES-OF-FREEDOM:
## Level of Significance = 0.05
##
## Call:
## gvlma(x = adiposelm1)
##
## Value p-value Decision
## Global Stat 1.372e+00 0.8491 Assumptions acceptable.
## Skewness 1.174e+00 0.2786 Assumptions acceptable.
## Kurtosis 3.779e-02 0.8459 Assumptions acceptable.
## Link Function 3.133e-15 1.0000 Assumptions acceptable.
## Heteroscedasticity 1.598e-01 0.6893 Assumptions acceptable.##
## Shapiro-Wilk normality test
##
## data: rstandard(adiposelm1)
## W = 0.97688, p-value = 0.4704## Warning in cor.test.default(fitted(adiposelm1), abs(rstandard(adiposelm1)), :
## Cannot compute exact p-value with ties##
## Spearman's rank correlation rho
##
## data: fitted(adiposelm1) and abs(rstandard(adiposelm1))
## S = 14364, p-value = 0.2547
## alternative hypothesis: true rho is not equal to 0
## sample estimates:
## rho
## 0.1695033## lag Autocorrelation D-W Statistic p-value
## 1 -0.1135751 2.212731 0.916
## Alternative hypothesis: rho != 0## No Studentized residuals with Bonferroni p < 0.05
## Largest |rstudent|:
## rstudent unadjusted p-value Bonferroni p
## 13 2.584726 0.013403 0.62992## Levene's Test for Homogeneity of Variance (center = median)
## Df F value Pr(>F)
## group 4 0.5104 0.7284
## 42Two-way factorial ANOVA example
R in Action 9.5, ToothGrowth dataset
This example uses the ToothGrowth dataset in the base installation to demonstrate a two-way between-groups ANOVA. Sixty guinea pigs are randomly assigned to receive one of three levels of ascorbic acid (0.5, 1, or 2mg), and one of two delivery methods (orange juice or Vitamin C), under the restriction that each treatment combination has 10 guinea pigs. The dependent variable is tooth length.
Load dataset
## dose
## supp 0.5 1 2
## OJ 10 10 10
## VC 10 10 10## Group.1 Group.2 x
## 1 OJ 0.5 13.23
## 2 VC 0.5 7.98
## 3 OJ 1.0 22.70
## 4 VC 1.0 16.77
## 5 OJ 2.0 26.06
## 6 VC 2.0 26.14## Group.1 Group.2 x
## 1 OJ 0.5 4.459709
## 2 VC 0.5 2.746634
## 3 OJ 1.0 3.910953
## 4 VC 1.0 2.515309
## 5 OJ 2.0 2.655058
## 6 VC 2.0 4.797731Perform factorial analysis of variance
## Analysis of Variance Table
##
## Response: len
## Df Sum Sq Mean Sq F value Pr(>F)
## supp 1 205.35 205.35 12.3170 0.0008936 ***
## dose 1 2224.30 2224.30 133.4151 < 2.2e-16 ***
## supp:dose 1 88.92 88.92 5.3335 0.0246314 *
## Residuals 56 933.63 16.67
## ---
## Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1Perform regression diagnostics and test assumptions
##
## Call:
## lm(formula = len ~ supp * dose)
##
## Residuals:
## Min 1Q Median 3Q Max
## -8.2264 -2.8462 0.0504 2.2893 7.9386
##
## Coefficients:
## Estimate Std. Error t value Pr(>|t|)
## (Intercept) 11.550 1.581 7.304 1.09e-09 ***
## suppVC -8.255 2.236 -3.691 0.000507 ***
## dose 7.811 1.195 6.534 2.03e-08 ***
## suppVC:dose 3.904 1.691 2.309 0.024631 *
## ---
## Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
##
## Residual standard error: 4.083 on 56 degrees of freedom
## Multiple R-squared: 0.7296, Adjusted R-squared: 0.7151
## F-statistic: 50.36 on 3 and 56 DF, p-value: 6.521e-16
##
##
## ASSESSMENT OF THE LINEAR MODEL ASSUMPTIONS
## USING THE GLOBAL TEST ON 4 DEGREES-OF-FREEDOM:
## Level of Significance = 0.05
##
## Call:
## gvlma(x = fit)
##
## Value p-value Decision
## Global Stat 10.6646 0.030604 Assumptions NOT satisfied!
## Skewness 0.0934 0.759902 Assumptions acceptable.
## Kurtosis 1.0752 0.299778 Assumptions acceptable.
## Link Function 8.7812 0.003043 Assumptions NOT satisfied!
## Heteroscedasticity 0.7148 0.397854 Assumptions acceptable.##
## Shapiro-Wilk normality test
##
## data: rstandard(fit)
## W = 0.98147, p-value = 0.4936## Warning in cor.test.default(fitted(fit), abs(rstandard(fit)), method =
## "spearman"): Cannot compute exact p-value with ties##
## Spearman's rank correlation rho
##
## data: fitted(fit) and abs(rstandard(fit))
## S = 35096, p-value = 0.8506
## alternative hypothesis: true rho is not equal to 0
## sample estimates:
## rho
## 0.02483306## lag Autocorrelation D-W Statistic p-value
## 1 0.1333076 1.68846 0.09
## Alternative hypothesis: rho != 0## No Studentized residuals with Bonferroni p < 0.05
## Largest |rstudent|:
## rstudent unadjusted p-value Bonferroni p
## 22 -2.185495 0.033129 NA
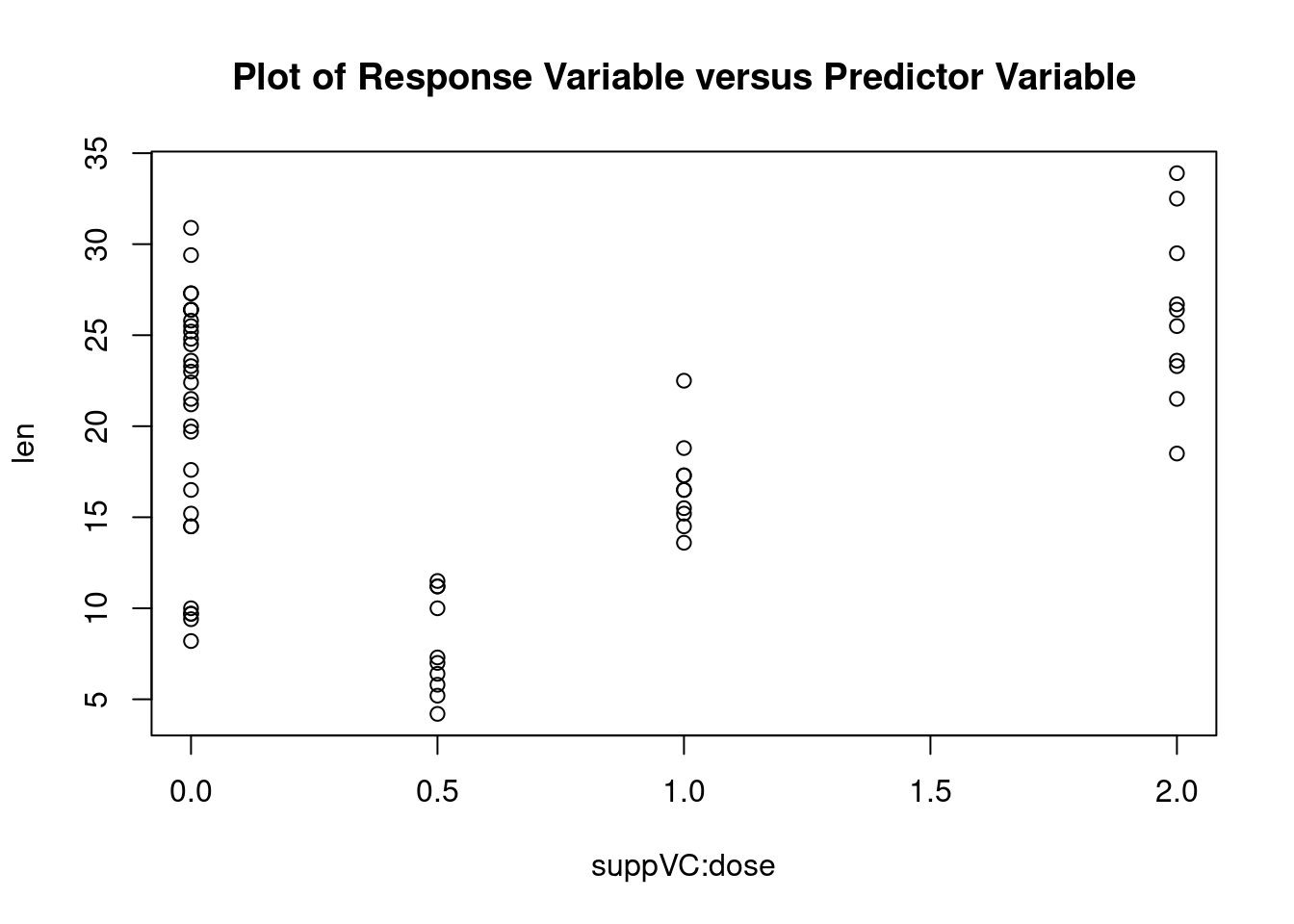
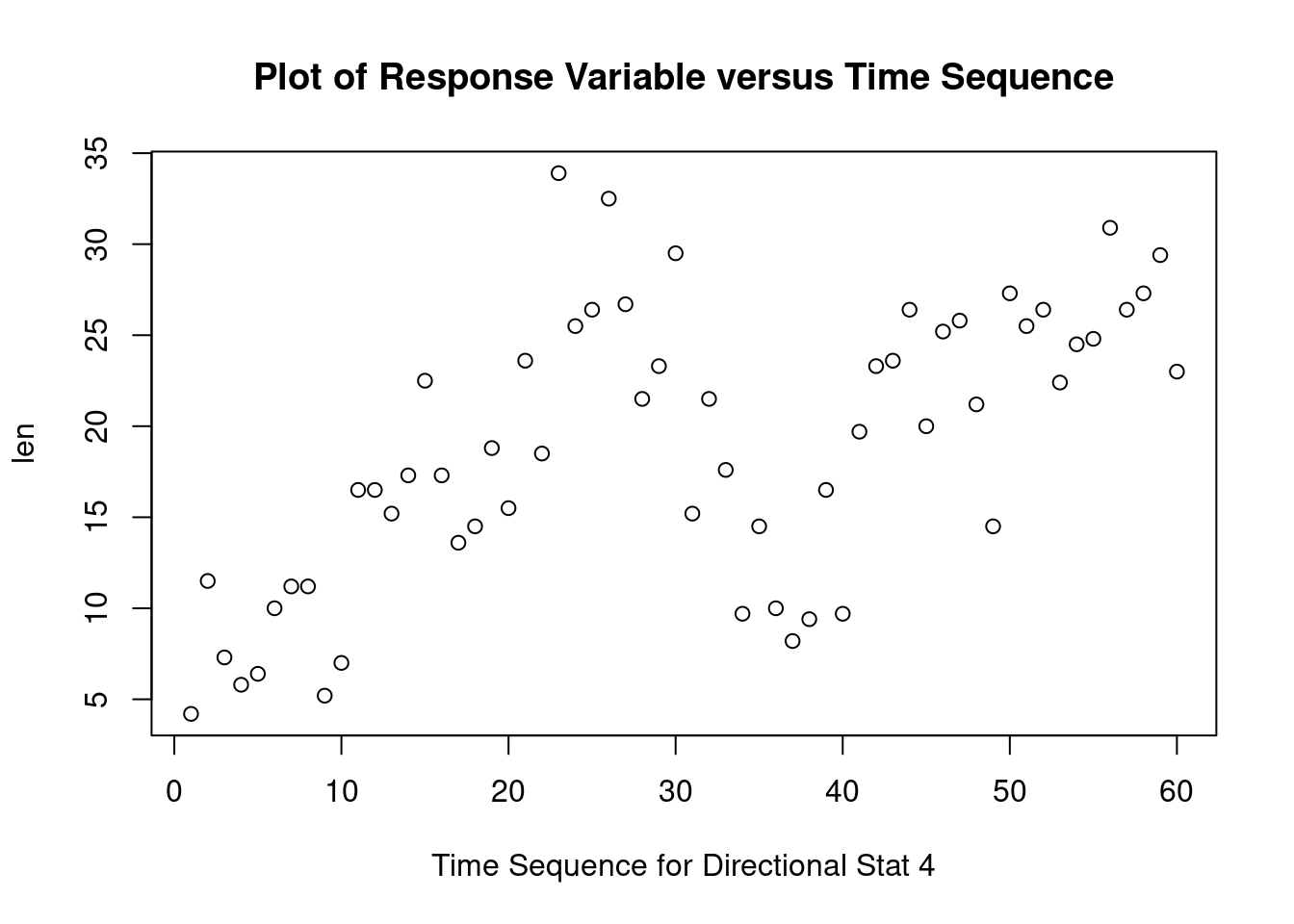
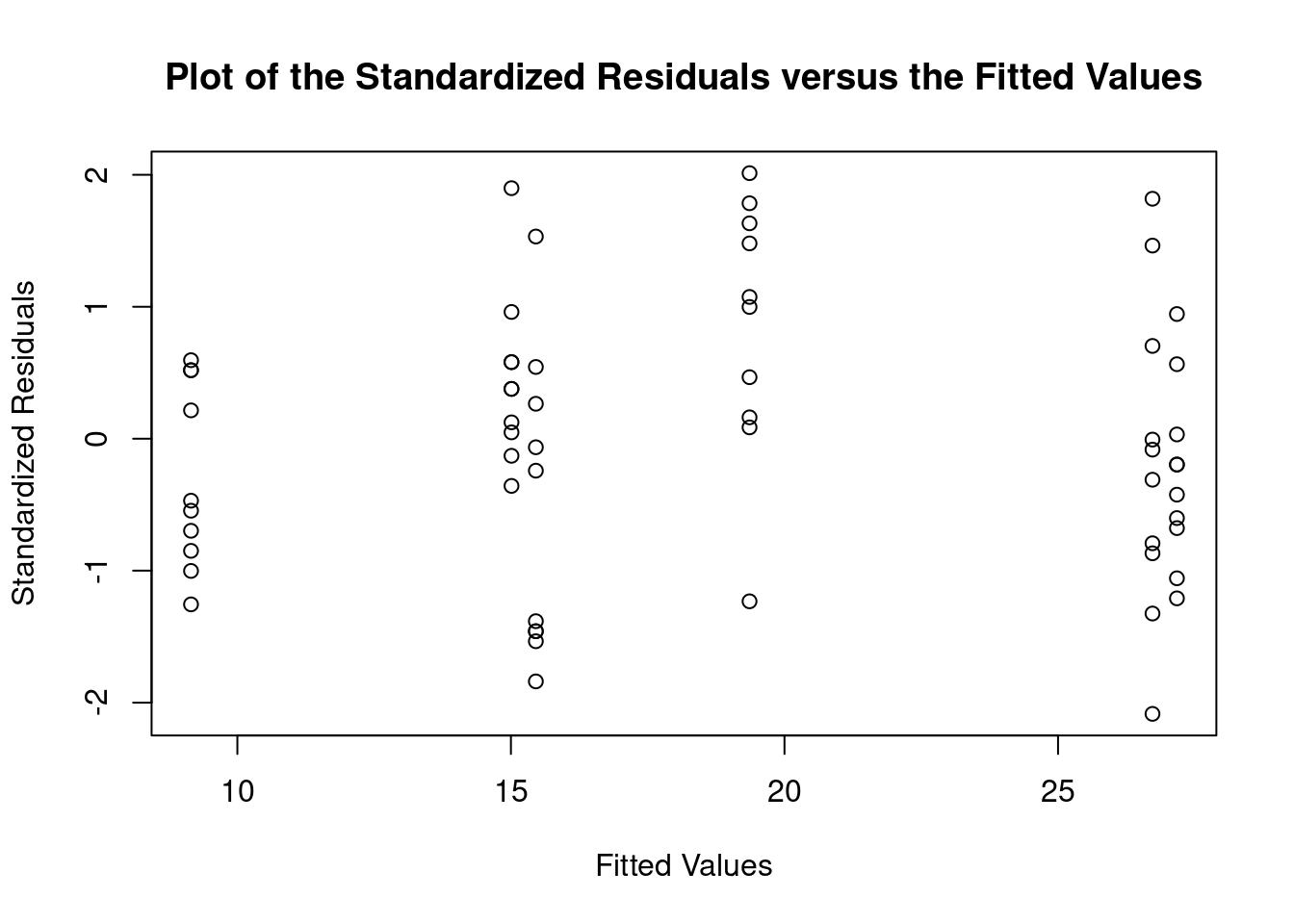
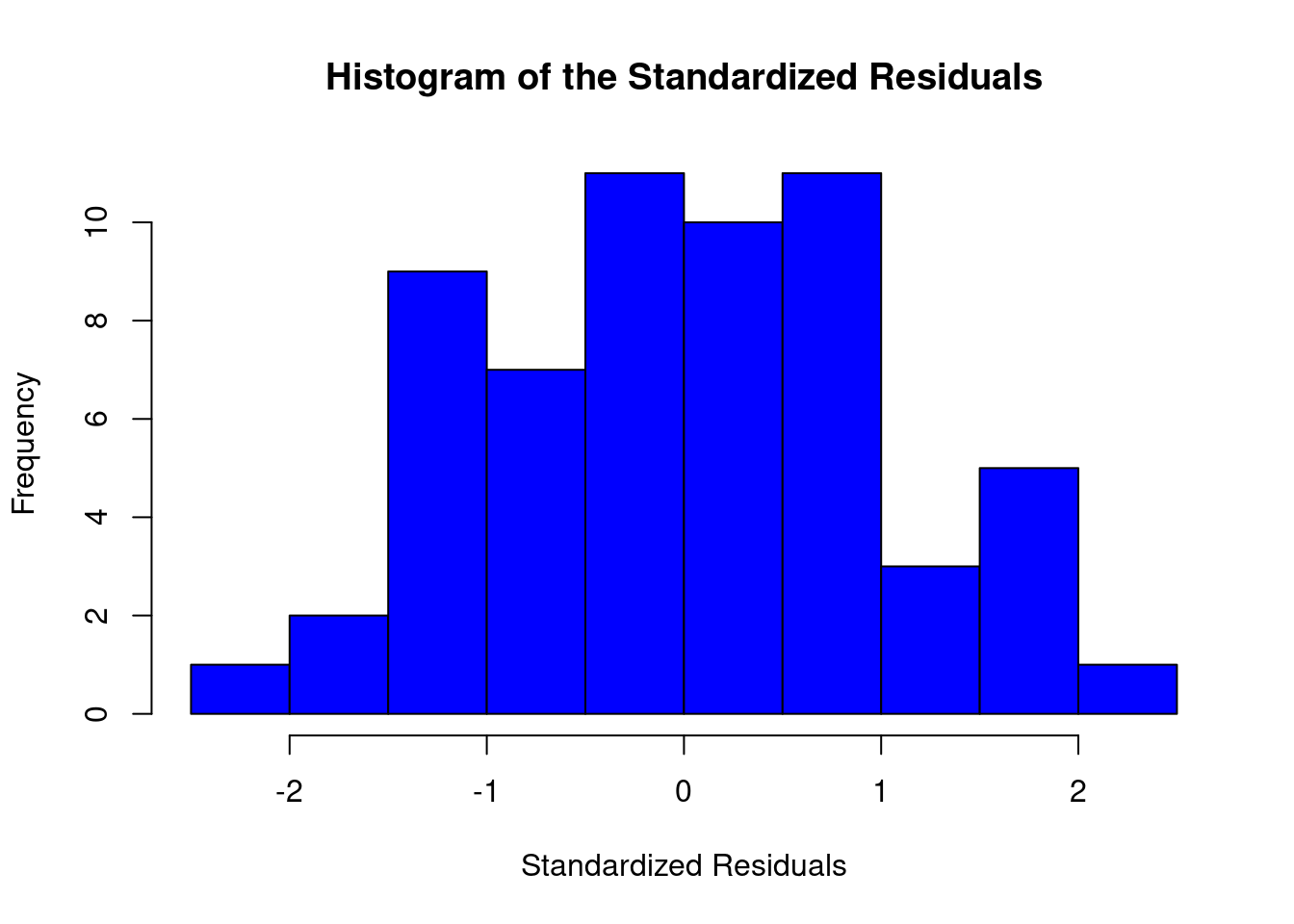
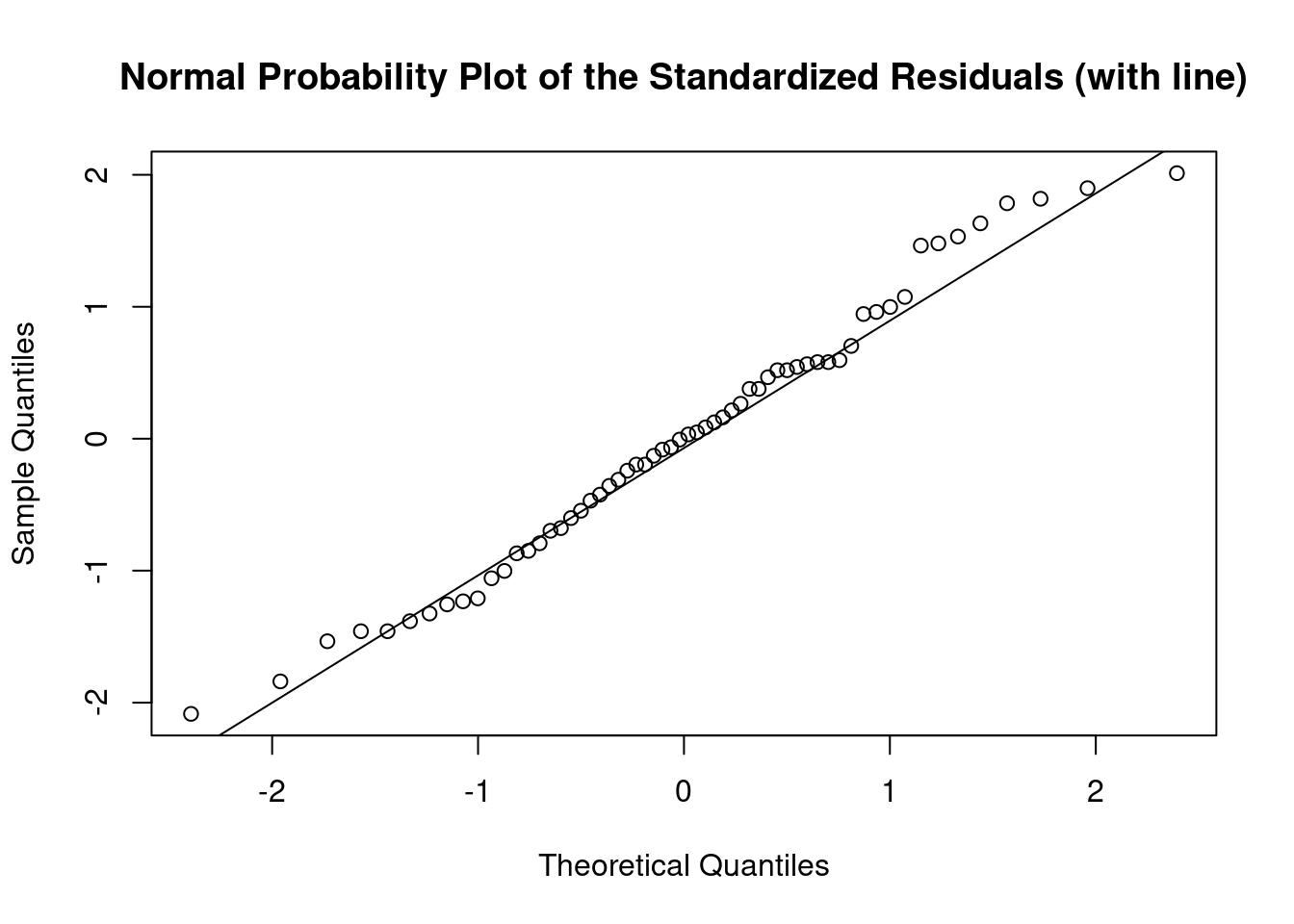
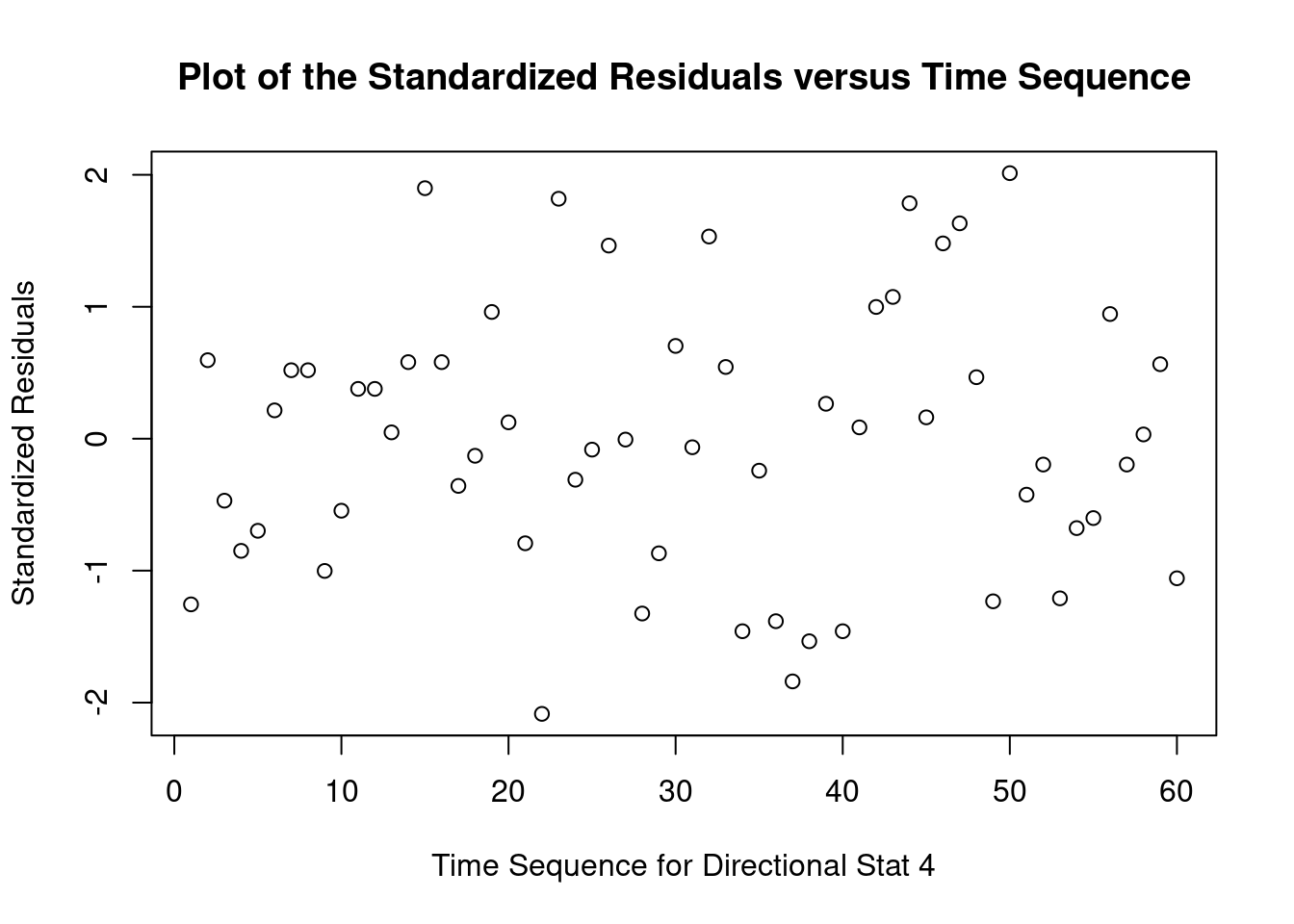
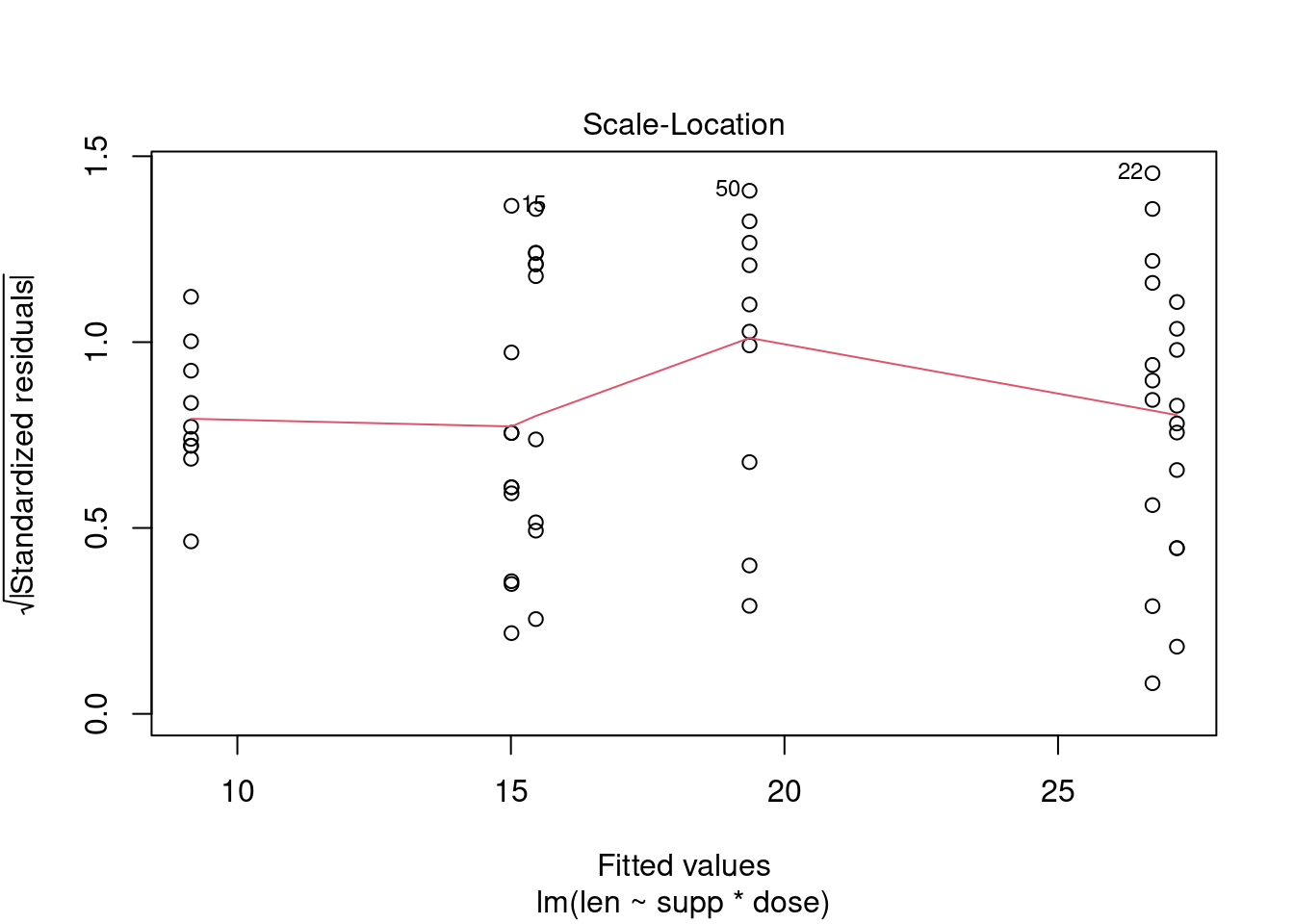
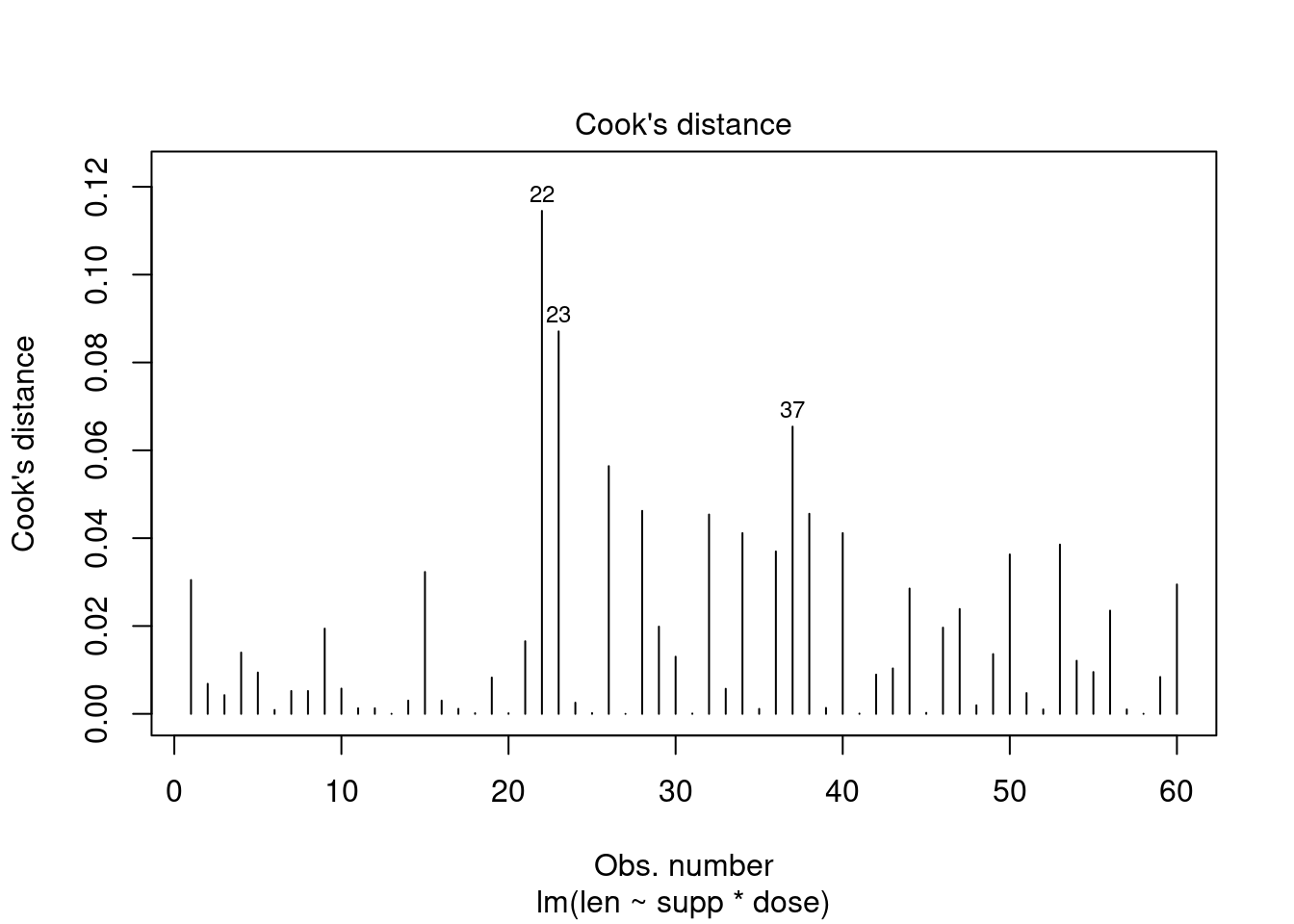
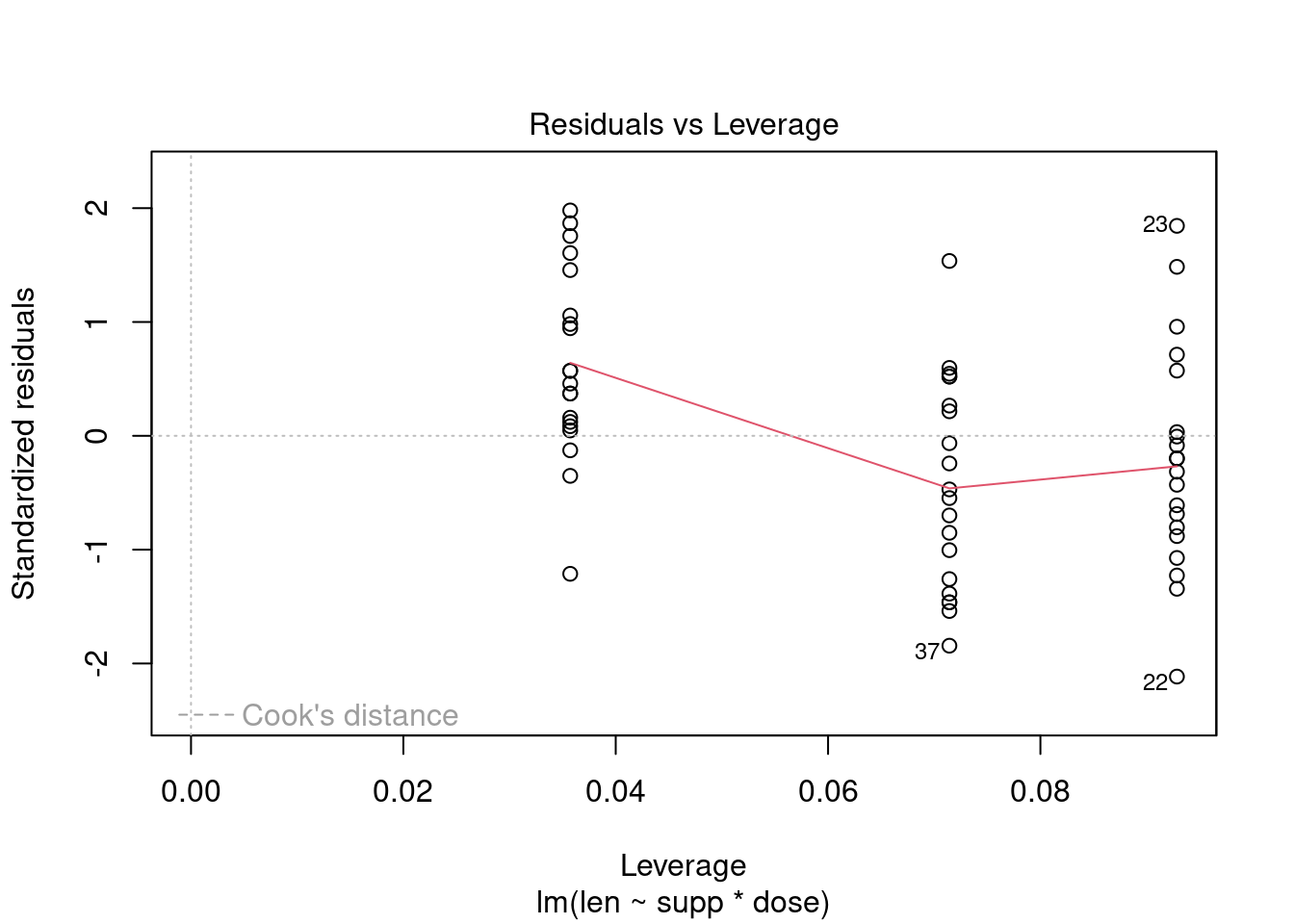
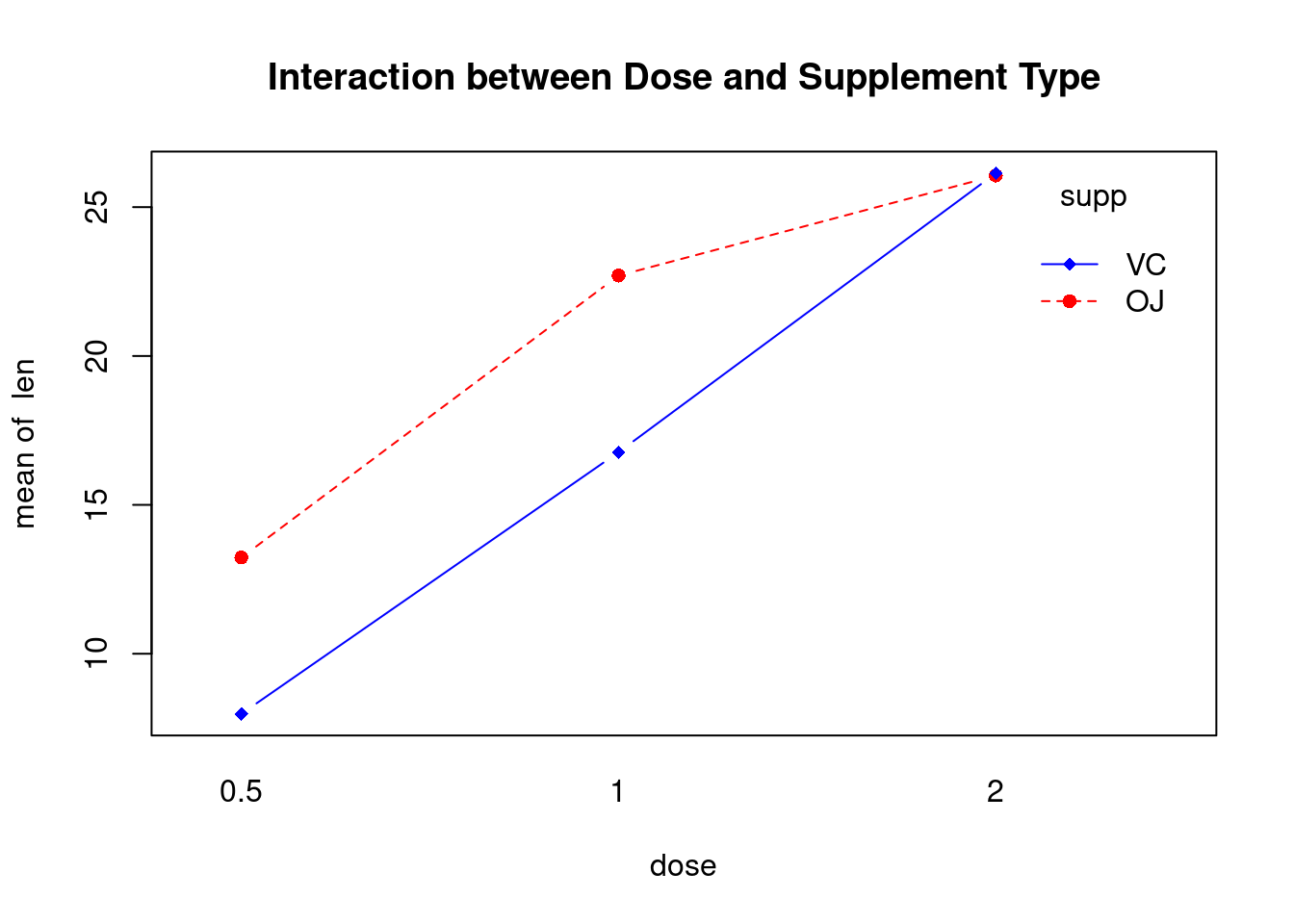
Two-way factorial ANOVA example
Atheroma thickness data from Smith BW et al, J Ultrasound Med 2012 (doi: 10.7863/jum.2012.31.5.711).
Import dataset
fall2009thickness <-
read.table(
'data/anova_factorial_jum2012_atheroma_thickness.csv',
header = TRUE,
sep = ','
)
attach(fall2009thickness)
head(fall2009thickness)## id uca mpa thickness
## 1 L604 Definity 0 35
## 2 L611 Definity 0 10
## 3 L614 Definity 0 70
## 4 L617 Definity 0 60
## 5 L624 Definity 0 100
## 6 L593 Saline 0 50Perform factorial analysis of variance
## Analysis of Variance Table
##
## Response: thickness
## Df Sum Sq Mean Sq F value Pr(>F)
## uca 1 9619 9618.9 4.6436 0.03775 *
## mpa 1 5255 5255.4 2.5371 0.11971
## uca:mpa 1 3285 3285.1 1.5859 0.21580
## Residuals 37 76643 2071.4
## ---
## Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1Perform regression diagnostics and test assumptions
##
## Call:
## lm(formula = thickness ~ uca * mpa, data = fall2009thickness)
##
## Residuals:
## Min 1Q Median 3Q Max
## -92.81 -26.34 -11.70 13.76 141.39
##
## Coefficients:
## Estimate Std. Error t value Pr(>|t|)
## (Intercept) 49.026 16.802 2.918 0.00596 **
## ucaSaline 7.320 23.838 0.307 0.76053
## mpa 3.608 12.762 0.283 0.77898
## ucaSaline:mpa 23.278 18.484 1.259 0.21580
## ---
## Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
##
## Residual standard error: 45.51 on 37 degrees of freedom
## Multiple R-squared: 0.1915, Adjusted R-squared: 0.126
## F-statistic: 2.922 on 3 and 37 DF, p-value: 0.04662
##
##
## ASSESSMENT OF THE LINEAR MODEL ASSUMPTIONS
## USING THE GLOBAL TEST ON 4 DEGREES-OF-FREEDOM:
## Level of Significance = 0.05
##
## Call:
## gvlma(x = thick_fit)
##
## Value p-value Decision
## Global Stat 15.0581 0.004582 Assumptions NOT satisfied!
## Skewness 5.2041 0.022534 Assumptions NOT satisfied!
## Kurtosis 4.1197 0.042388 Assumptions NOT satisfied!
## Link Function 0.9562 0.328138 Assumptions acceptable.
## Heteroscedasticity 4.7782 0.028823 Assumptions NOT satisfied!##
## Shapiro-Wilk normality test
##
## data: rstandard(thick_fit)
## W = 0.95015, p-value = 0.07084## Warning in cor.test.default(fitted(thick_fit), abs(rstandard(thick_fit)), :
## Cannot compute exact p-value with ties##
## Spearman's rank correlation rho
##
## data: fitted(thick_fit) and abs(rstandard(thick_fit))
## S = 9194.5, p-value = 0.2121
## alternative hypothesis: true rho is not equal to 0
## sample estimates:
## rho
## 0.199083## lag Autocorrelation D-W Statistic p-value
## 1 -0.3832382 2.76177 0.04
## Alternative hypothesis: rho != 0## rstudent unadjusted p-value Bonferroni p
## 27 3.699668 0.00071694 0.029395Generate plots
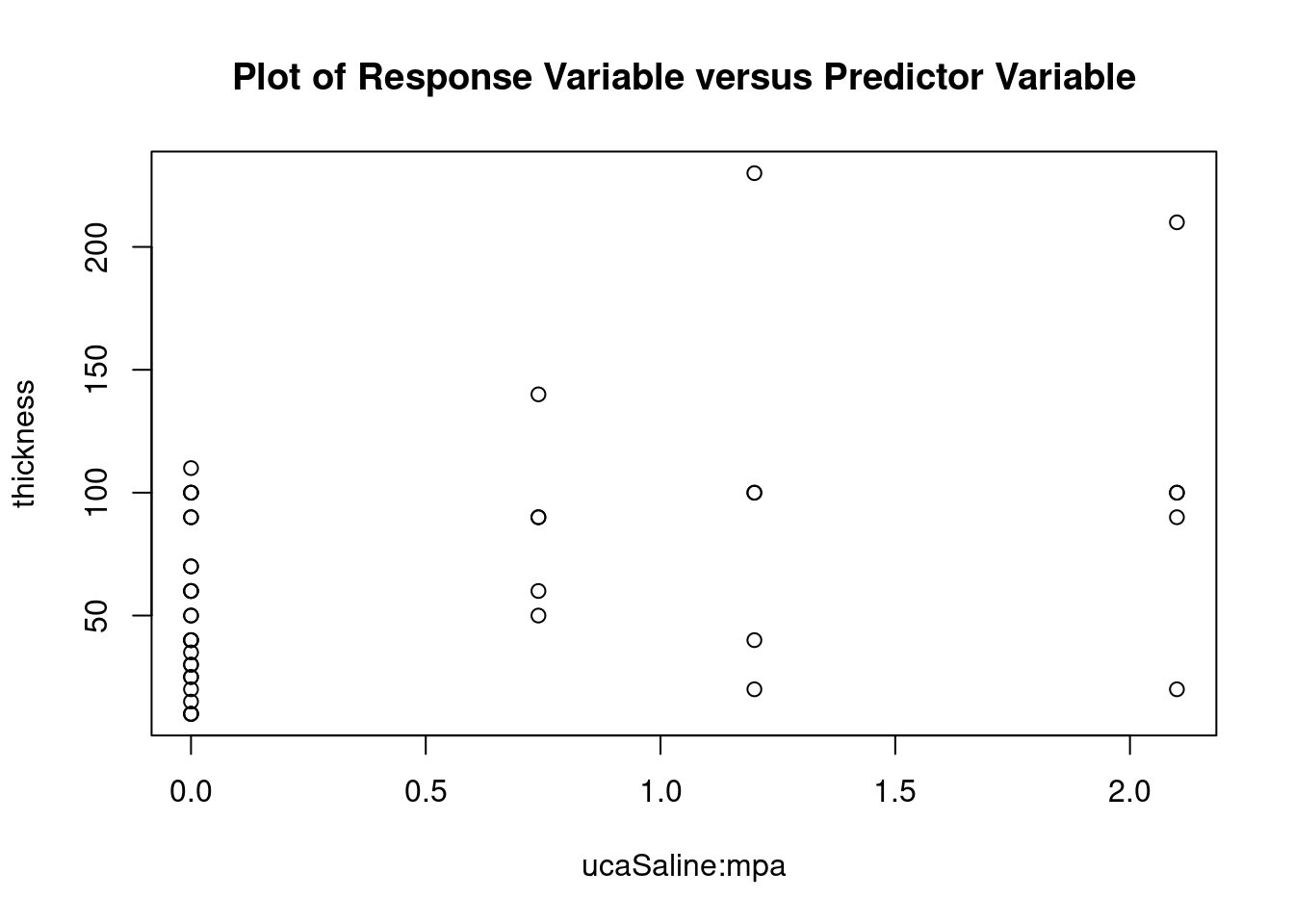
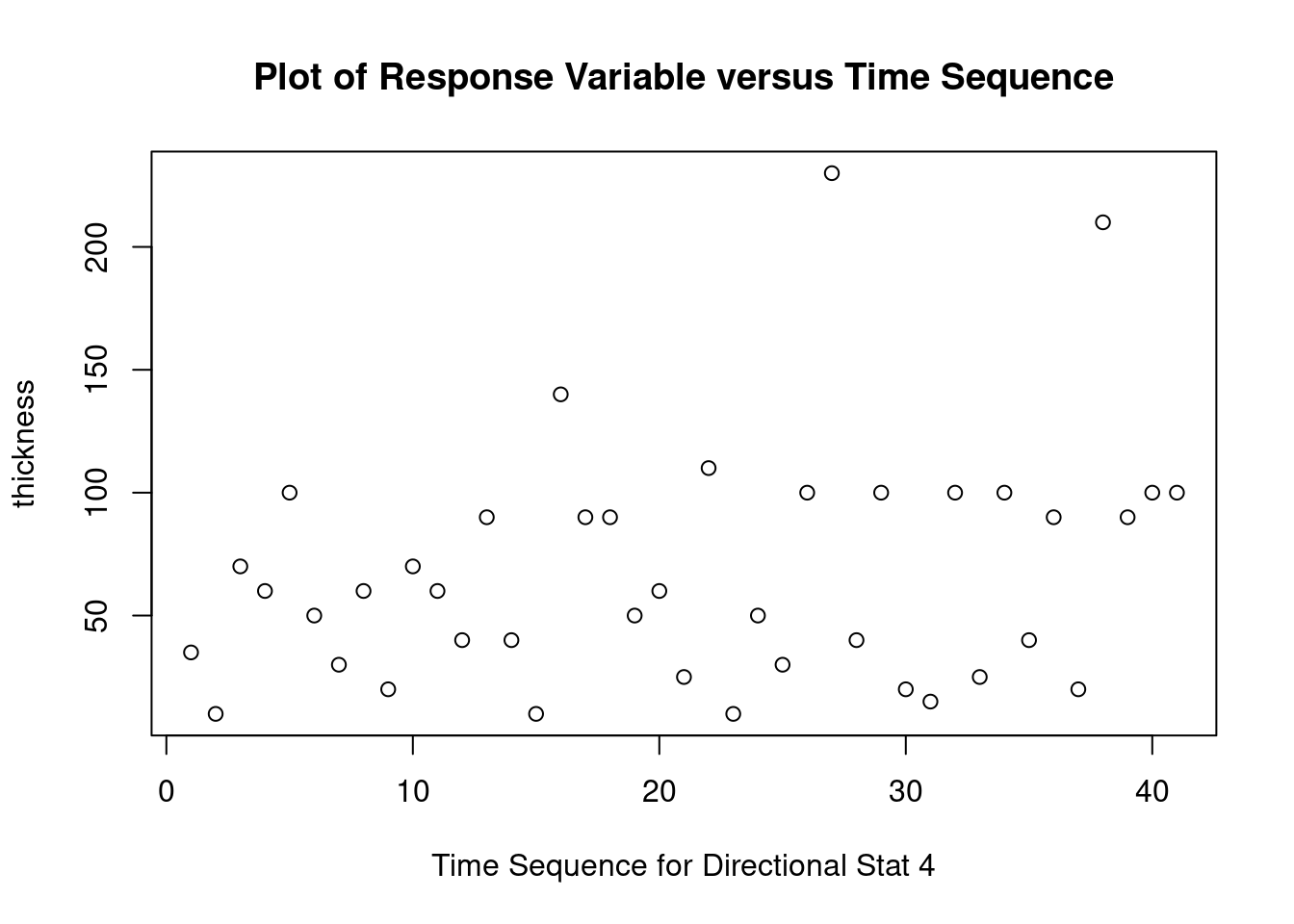
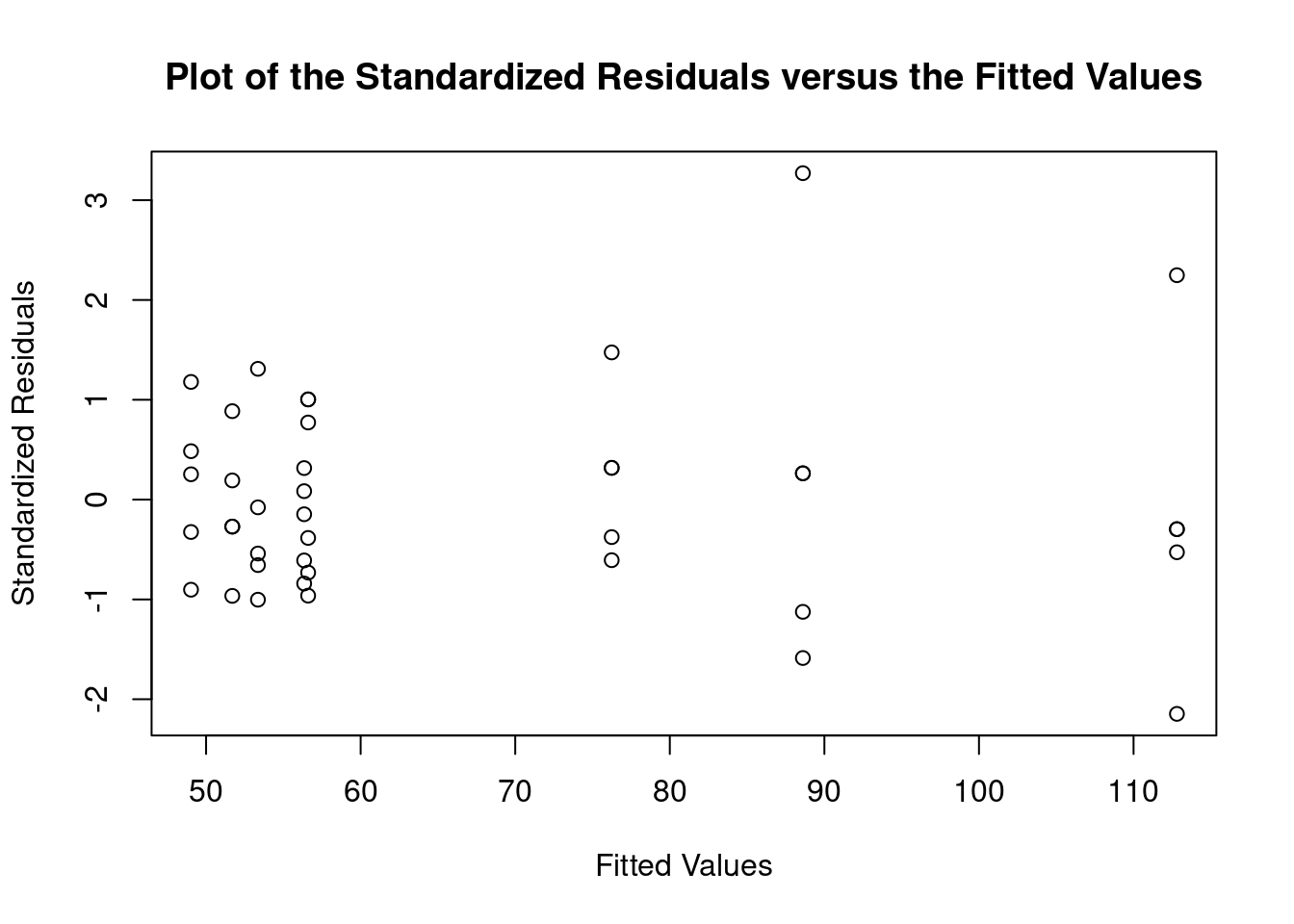
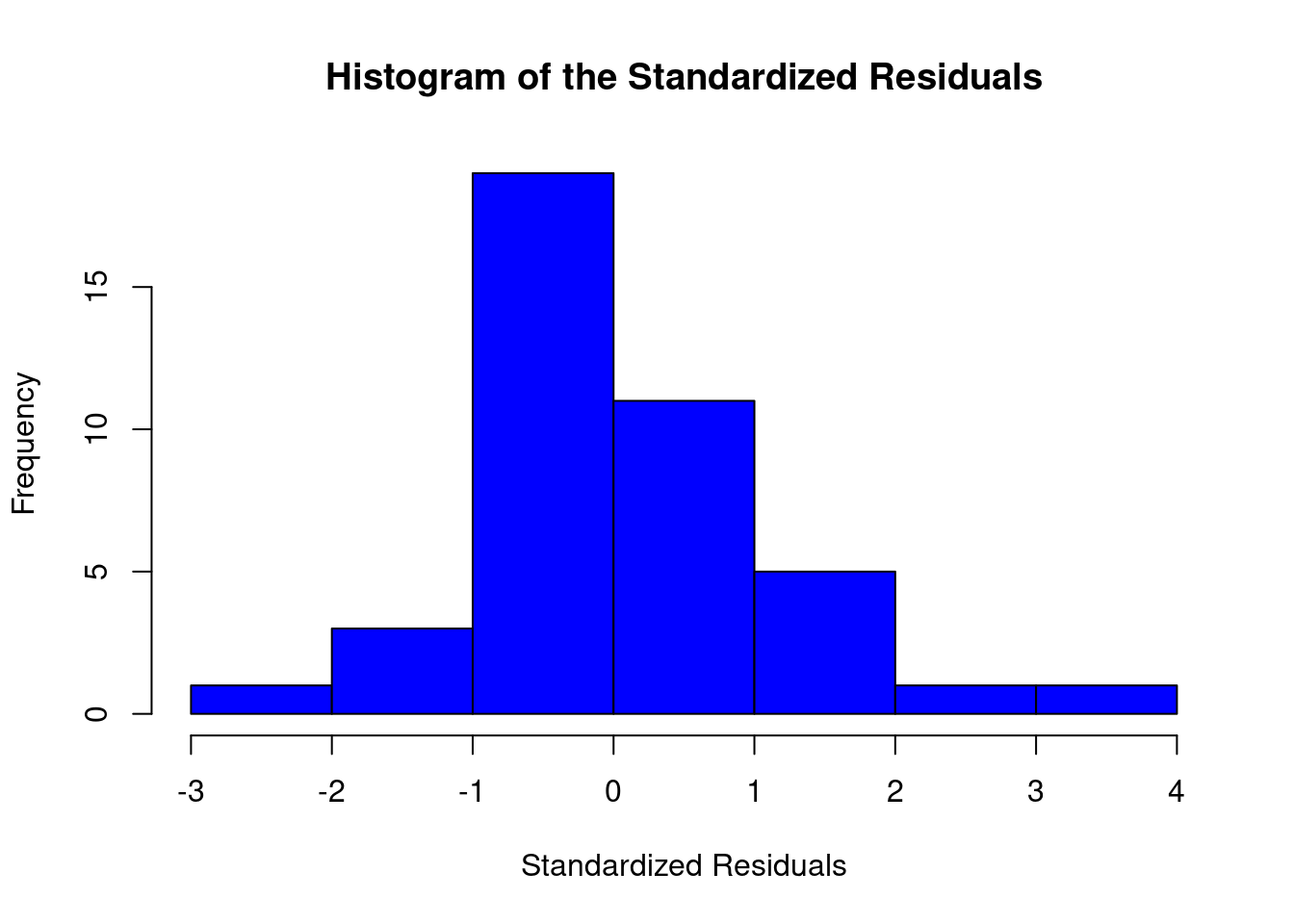
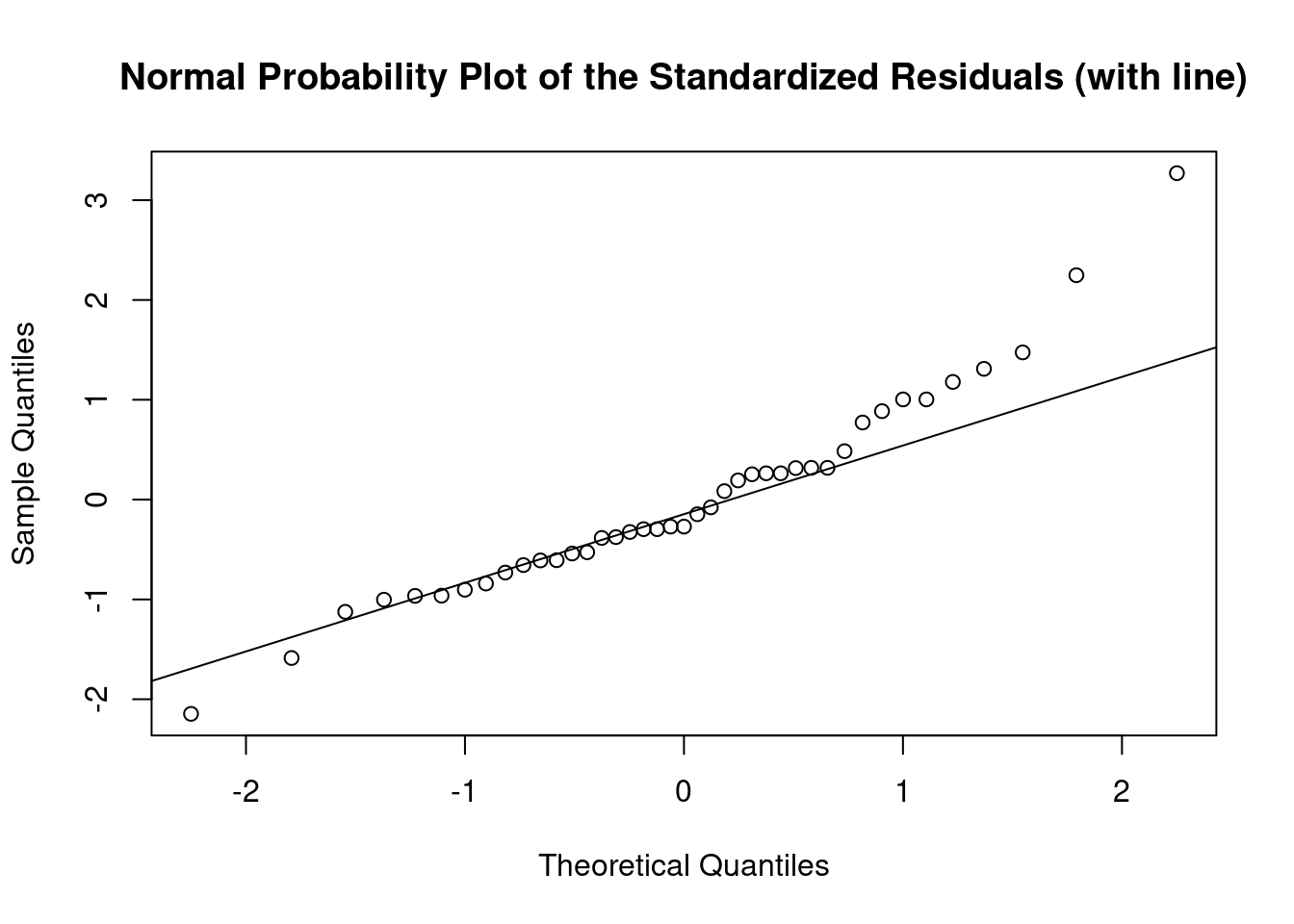
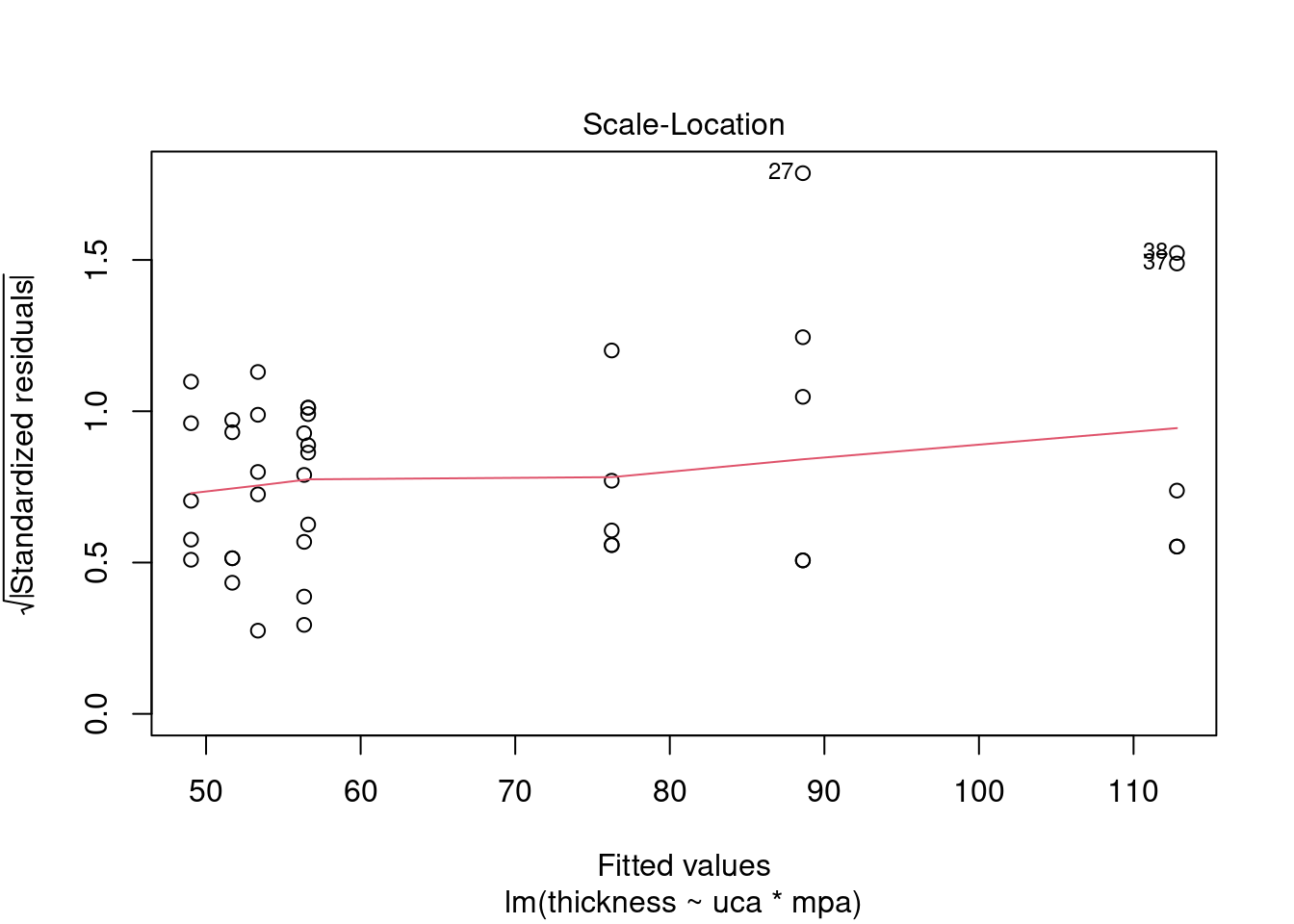
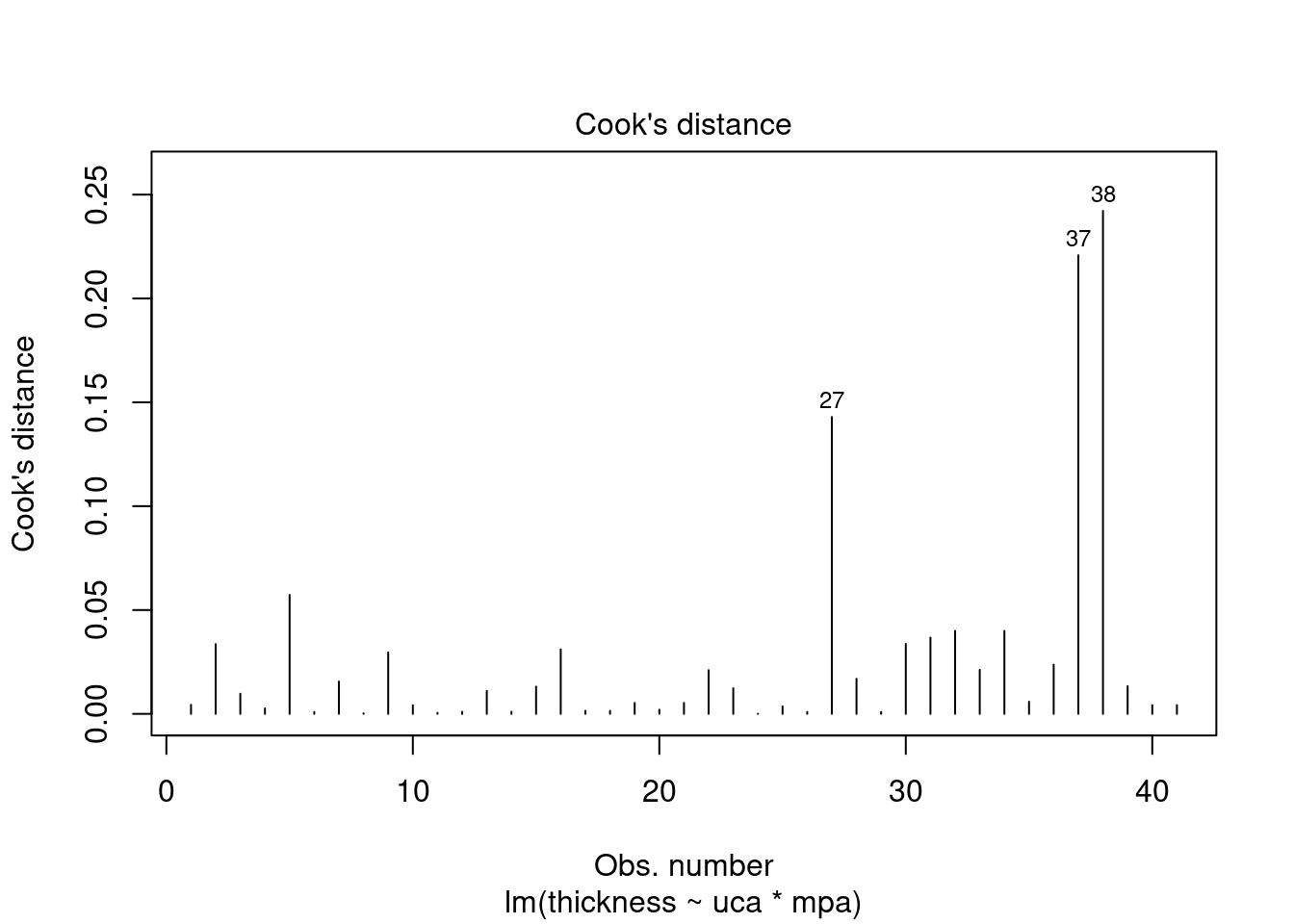
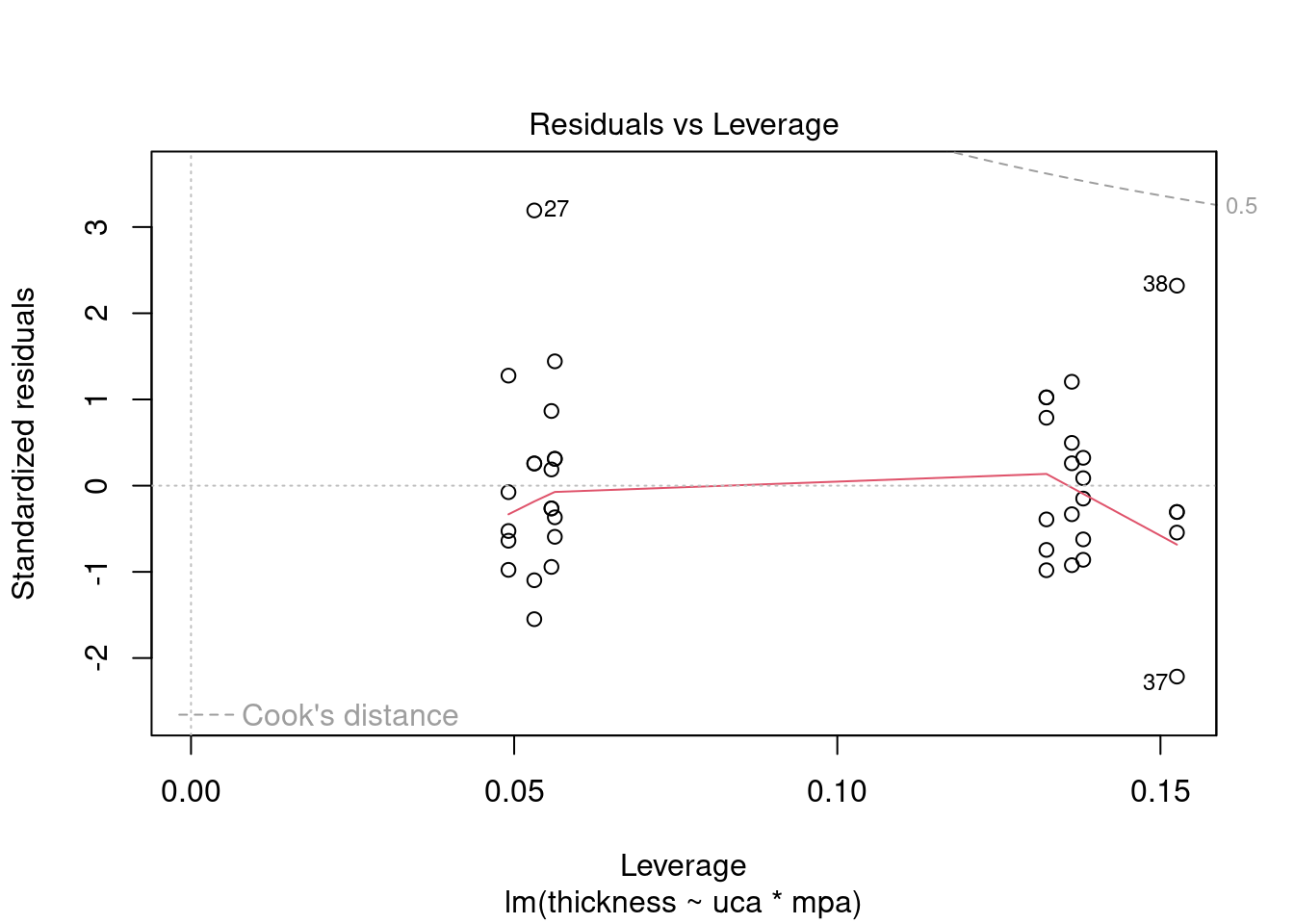
interaction.plot(
uca,
mpa,
thickness,
type = 'b',
col = c('red', 'blue'),
pch = c(16, 18),
main = 'Interaction between contrast agent and pressure level'
)
Log transform to correct non-normality, heteroscedasticity, and outlier
## Analysis of Variance Table
##
## Response: log(thickness)
## Df Sum Sq Mean Sq F value Pr(>F)
## uca 1 2.4372 2.43722 4.1806 0.04805 *
## mpa 1 0.6951 0.69511 1.1923 0.28192
## uca:mpa 1 0.3084 0.30843 0.5291 0.47158
## Residuals 37 21.5703 0.58298
## ---
## Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1Perform regression diagnostics and test assumptions
##
## Call:
## lm(formula = log(thickness) ~ uca * mpa, data = fall2009thickness)
##
## Residuals:
## Min 1Q Median 3Q Max
## -1.51653 -0.50113 0.09291 0.44825 1.18425
##
## Coefficients:
## Estimate Std. Error t value Pr(>|t|)
## (Intercept) 3.64610 0.28187 12.935 2.67e-15 ***
## ucaSaline 0.26316 0.39991 0.658 0.515
## mpa 0.06159 0.21410 0.288 0.775
## ucaSaline:mpa 0.22555 0.31009 0.727 0.472
## ---
## Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
##
## Residual standard error: 0.7635 on 37 degrees of freedom
## Multiple R-squared: 0.1376, Adjusted R-squared: 0.06764
## F-statistic: 1.967 on 3 and 37 DF, p-value: 0.1358
##
##
## ASSESSMENT OF THE LINEAR MODEL ASSUMPTIONS
## USING THE GLOBAL TEST ON 4 DEGREES-OF-FREEDOM:
## Level of Significance = 0.05
##
## Call:
## gvlma(x = log_thick_fit)
##
## Value p-value Decision
## Global Stat 3.4821 0.4806 Assumptions acceptable.
## Skewness 2.1479 0.1428 Assumptions acceptable.
## Kurtosis 0.5245 0.4689 Assumptions acceptable.
## Link Function 0.6395 0.4239 Assumptions acceptable.
## Heteroscedasticity 0.1701 0.6800 Assumptions acceptable.##
## Shapiro-Wilk normality test
##
## data: rstandard(log_thick_fit)
## W = 0.93909, p-value = 0.02924## Warning in cor.test.default(fitted(log_thick_fit),
## abs(rstandard(log_thick_fit)), : Cannot compute exact p-value with ties##
## Spearman's rank correlation rho
##
## data: fitted(log_thick_fit) and abs(rstandard(log_thick_fit))
## S = 12485, p-value = 0.5861
## alternative hypothesis: true rho is not equal to 0
## sample estimates:
## rho
## -0.08758111## lag Autocorrelation D-W Statistic p-value
## 1 -0.3411439 2.681506 0.068
## Alternative hypothesis: rho != 0## No Studentized residuals with Bonferroni p < 0.05
## Largest |rstudent|:
## rstudent unadjusted p-value Bonferroni p
## 37 -2.276215 0.028882 NAANOVA Compact Letter Display
Compact Letter Display (CLD) for ANOVA multiple comparisons with the
multcomp package
# multiple comparison procedures
# set up a one-way ANOVA
library(multcomp)
data(warpbreaks)
amod <- aov(breaks ~ tension, data = warpbreaks)
# specify all pair-wise comparisons among levels of variable 'tension'
tuk <- glht(amod, linfct = mcp(tension = 'Tukey'))
# extract information
tuk.cld <- cld(tuk)
# use sufficiently large upper margin
old.par <- par(mai = c(1, 1, 1.5, 1))
# plot
plot(tuk.cld)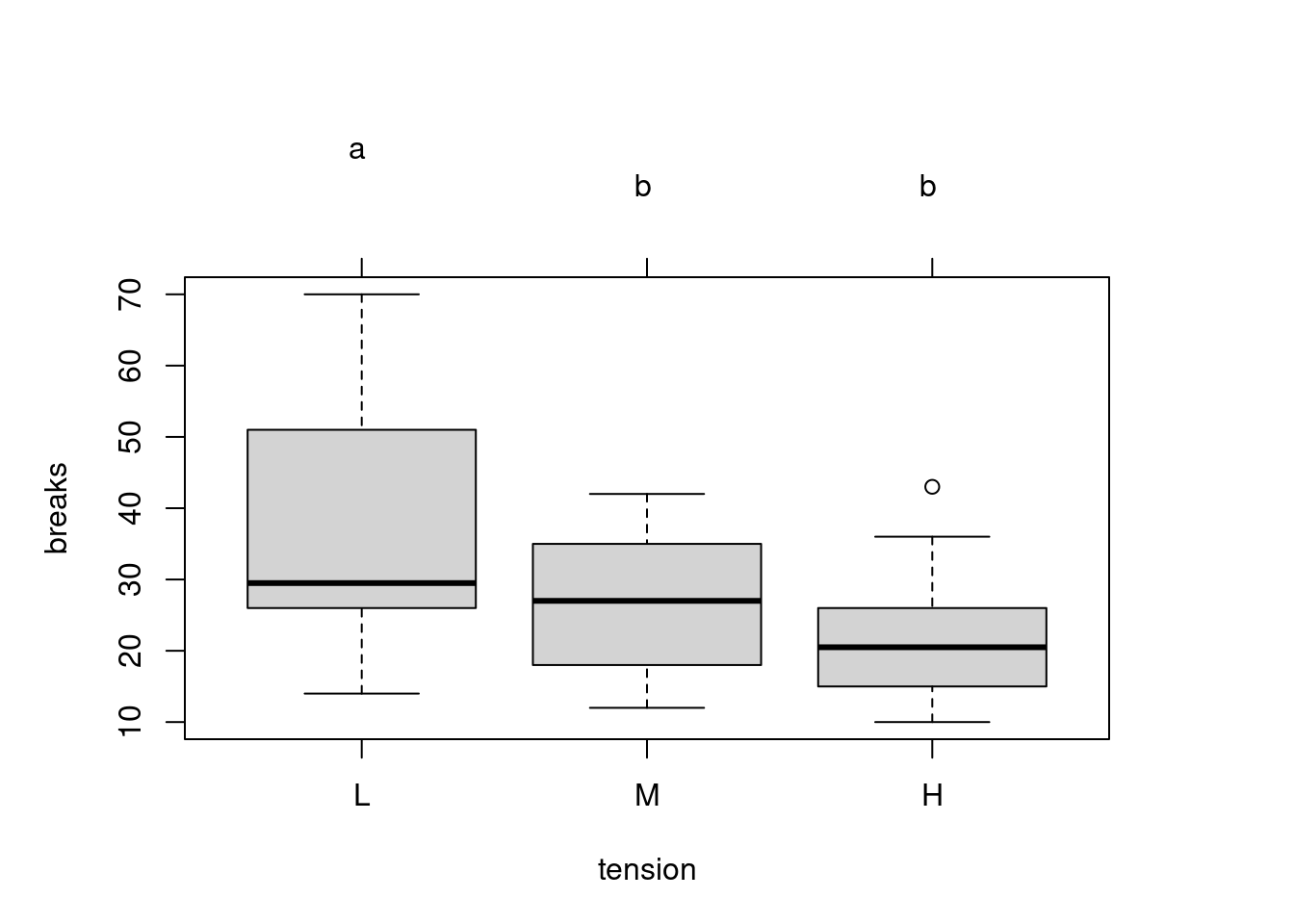
Repeated measures ANOVA
Repeated measures ANOVA example
R in Action 2nd ed, 9.6, CO2 dataset
note that the code from the first edition may return errors
In addition to the base installation, we’ll be using the
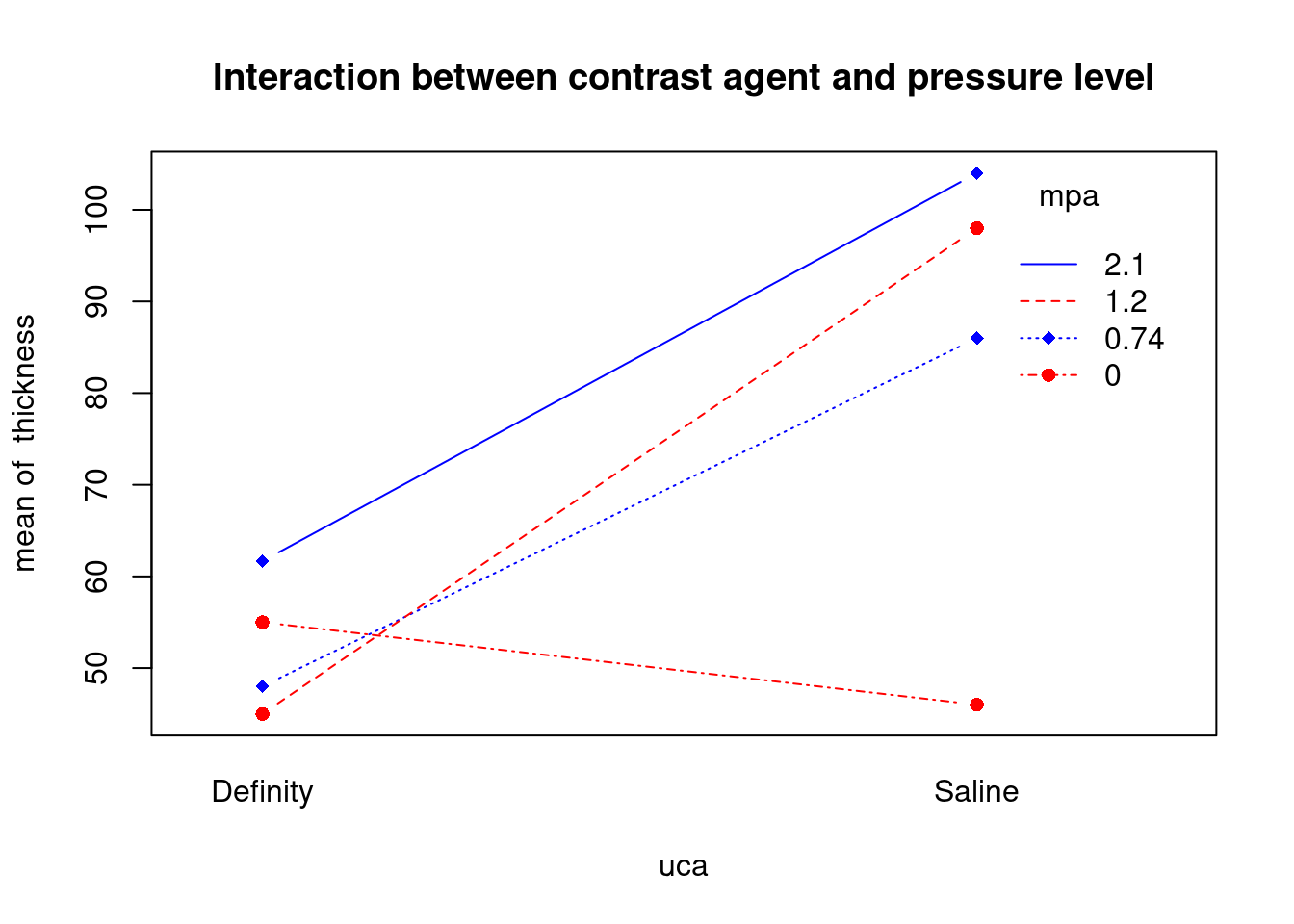
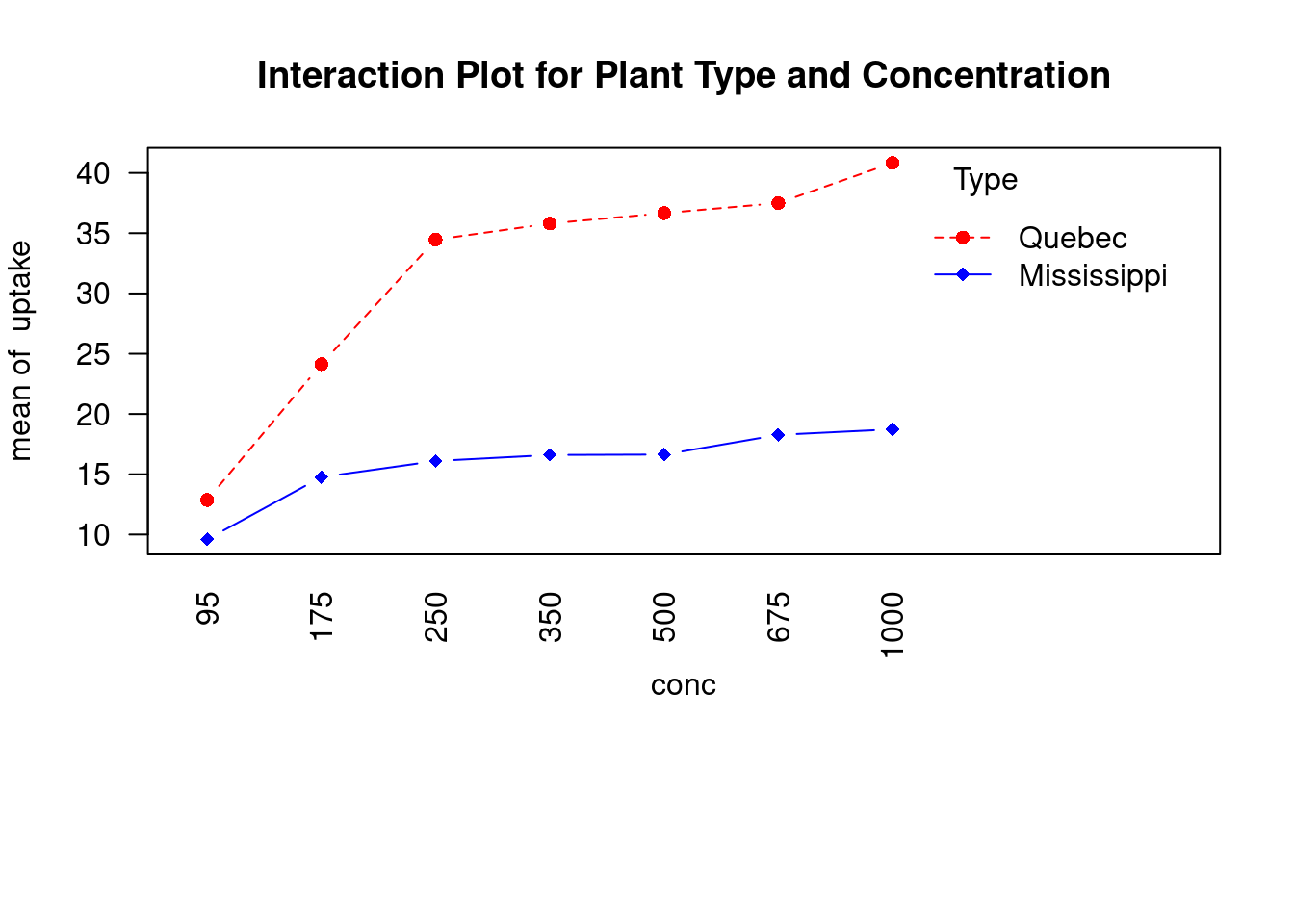
car,gplots,HH,rrcov, andmvoutlierpackages in our examples. Be sure to install them before trying out the sample code.The CO2 dataset included in the base installation contains the results of a study of cold tolerance in Northern and Southern plants of the grass species Echinochloa crus-galli (Potvin, Lechowicz, & Tardif, 1990). The photosynthetic rates of chilled plants were compared with the photosynthetic rates of nonchilled plants at several ambient CO2 concentrations. Half the plants were from Quebec, and half were from Mississippi.
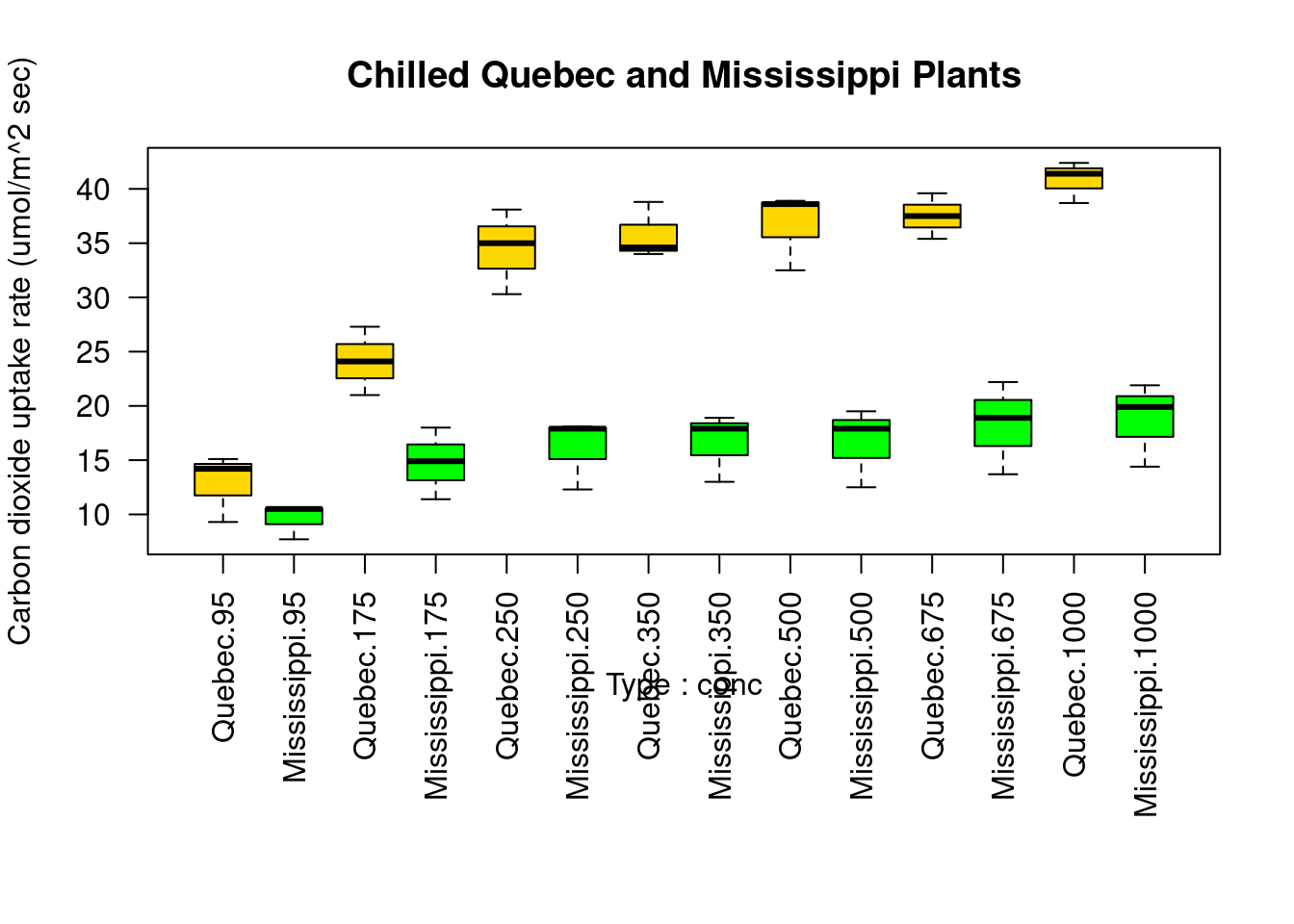
In this example, we’ll focus on chilled plants. The dependent variable is carbon dioxide uptake (uptake) in ml/L, and the independent variables are Type (Quebec versus Mississippi) and ambient CO2 concentration (conc) with seven levels (ranging from 95 to 1000 umol/m^2 sec). Type is a between-groups factor, and conc is a within- groups factor. Type is already stored as a factor, but you’ll need to convert conc to a factor before continuing. The analysis is presented in the next listing.
CO2$conc <- factor(CO2$conc)
w1b1 <- subset(CO2, Treatment == 'chilled')
co2_fit <- aov(uptake ~ conc * Type + Error(Plant / (conc)), w1b1)
summary(co2_fit)##
## Error: Plant
## Df Sum Sq Mean Sq F value Pr(>F)
## Type 1 2667.2 2667.2 60.41 0.00148 **
## Residuals 4 176.6 44.1
## ---
## Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
##
## Error: Plant:conc
## Df Sum Sq Mean Sq F value Pr(>F)
## conc 6 1472.4 245.40 52.52 1.26e-12 ***
## conc:Type 6 428.8 71.47 15.30 3.75e-07 ***
## Residuals 24 112.1 4.67
## ---
## Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1par(las = 2)
par(mar = c(10, 4, 4, 2))
with(
w1b1,
interaction.plot(
conc,
Type,
uptake,
type = 'b',
col = c('red', 'blue'),
pch = c(16, 18),
main = 'Interaction Plot for Plant Type and Concentration'
)
)
boxplot(
uptake ~ Type * conc,
data = w1b1,
col = (c('gold', 'green')),
main = 'Chilled Quebec and Mississippi Plants',
ylab = 'Carbon dioxide uptake rate (umol/m^2 sec)'
)
The many approaches to mixed-model designs
The CO2 example in this section was analyzed using a traditional repeated measures ANOVA. The approach assumes that the covariance matrix for any within-groups factor follows a specified form known as sphericity. Specifically, it assumes that the vari- ances of the differences between any two levels of the within-groups factor are equal. In real-world data, it’s unlikely that this assumption will be met. This has led to a number of alternative approaches, including the following:
- Using the lmer() function in the lme4 package to fit linear mixed models (Bates, 2005)
- Using the Anova() function in the car package to adjust traditional test statis- tics to account for lack of sphericity (for example, the Geisser–Greenhouse correction)
- Using the gls() function in the nlme package to fit generalized least squares models with specified variance-covariance structures (UCLA, 2009)
- Using multivariate analysis of variance to model repeated measured data (Hand, 1987)
Coverage of these approaches is beyond the scope of this text. If you’re interested in learning more, check out Pinheiro and Bates (2000) and Zuur et al. (2009).
ANCOVA
One-way ANCOVA example
R in Action 9.4, litter dataset from multcomp package
Pregnant mice were divided into four treatment groups; each group received a different dose of a drug (0, 5, 50, or 500). The mean post-birth weight for each litter was the dependent variable and gestation time was included as a covariate. The analysis is given in the following listing.
Load dataset
## dose
## 0 5 50 500
## 20 19 18 17## Group.1 x
## 1 0 32.30850
## 2 5 29.30842
## 3 50 29.86611
## 4 500 29.64647Perform analysis of covariance
## Df Sum Sq Mean Sq F value Pr(>F)
## gesttime 1 134.3 134.30 8.049 0.00597 **
## dose 3 137.1 45.71 2.739 0.04988 *
## Residuals 69 1151.3 16.69
## ---
## Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1Calculate adjusted group means
Because you’re using a covariate, you may want to obtain adjusted group means—that is, the group means obtained after partialing out the effects of the covariate. You can use the effect() function in the effects library to calculate adjusted means:
## lattice theme set by effectsTheme()
## See ?effectsTheme for details.##
## dose effect
## dose
## 0 5 50 500
## 32.35367 28.87672 30.56614 29.33460In this case, the adjusted means are similar to the unadjusted means produced by the aggregate() function, but this won’t always be the case. The effects package provides a powerful method of obtaining adjusted means for complex research designs and presenting them visually. See the package documentation on CRAN for more details.
Multiple comparisons
library(multcomp)
litter_contrast <- rbind('no drug vs. drug' = c(3,-1,-1,-1))
summary(glht(litter_fit, linfct = mcp(dose = litter_contrast)))##
## Simultaneous Tests for General Linear Hypotheses
##
## Multiple Comparisons of Means: User-defined Contrasts
##
##
## Fit: aov(formula = weight ~ gesttime + dose)
##
## Linear Hypotheses:
## Estimate Std. Error t value Pr(>|t|)
## no drug vs. drug == 0 8.284 3.209 2.581 0.012 *
## ---
## Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
## (Adjusted p values reported -- single-step method)The contrast c(3, -1, -1, -1) specifies a comparison of the first group with the average of the other three. The hypothesis is tested with a t statistic (2.581 in this case), which is significant at the p < .05 level. Therefore, you can conclude that the no-drug group has a higher birth weight than drug conditions. Other contrasts can be added to the rbind() function (see help(glht) for details).
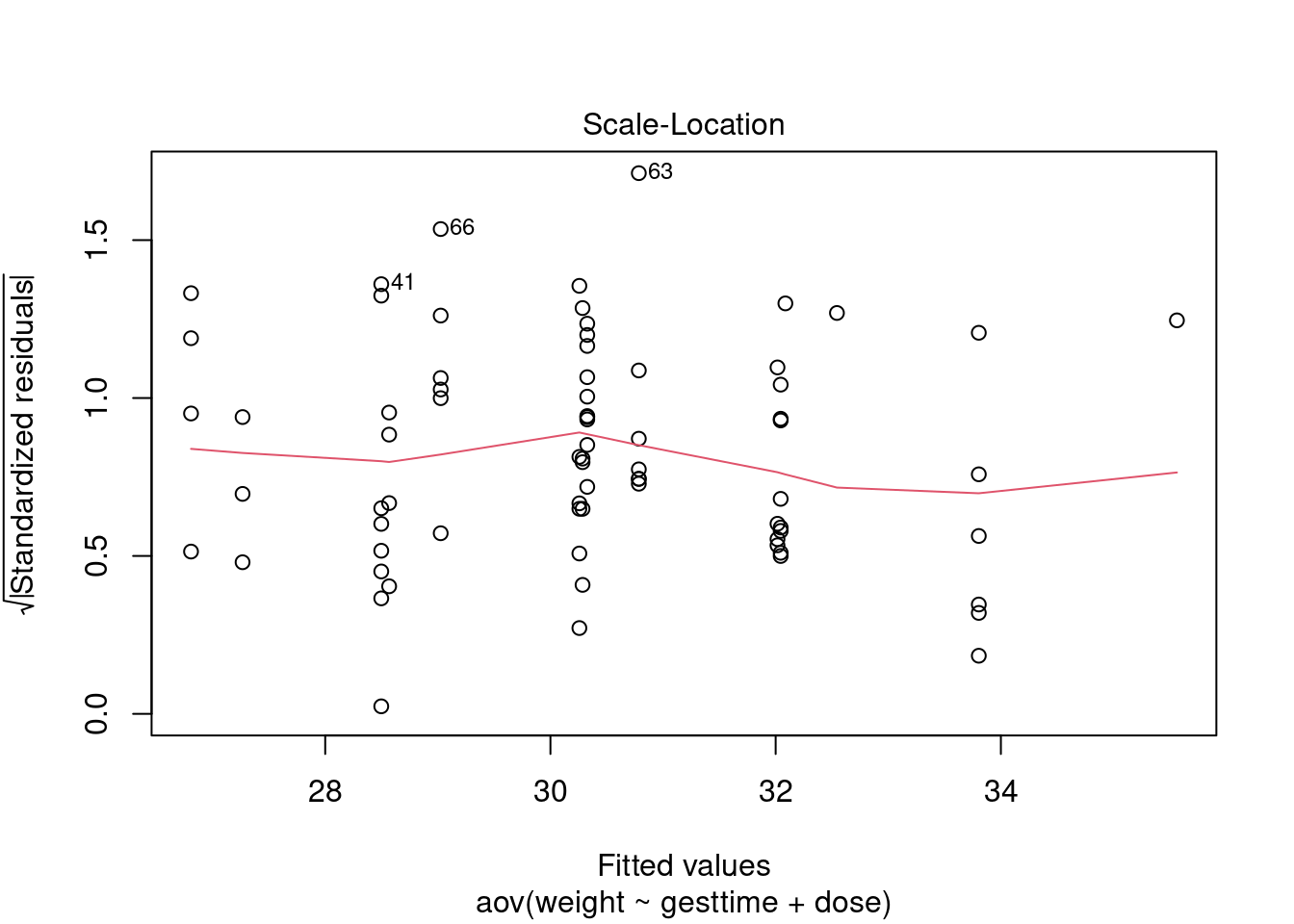
Assumptions
# global validation of linear model assumptions
library(gvlma)
gvlmalitter <- gvlma(lm(weight ~ gesttime + dose, data = litter))
summary(gvlmalitter)##
## Call:
## lm(formula = weight ~ gesttime + dose, data = litter)
##
## Residuals:
## Min 1Q Median 3Q Max
## -11.5649 -2.0068 0.1476 3.0755 7.3023
##
## Coefficients:
## Estimate Std. Error t value Pr(>|t|)
## (Intercept) -45.366 24.984 -1.816 0.07374 .
## gesttime 3.519 1.131 3.111 0.00271 **
## dose5 -3.477 1.318 -2.639 0.01027 *
## dose50 -1.788 1.344 -1.330 0.18780
## dose500 -3.019 1.352 -2.232 0.02883 *
## ---
## Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
##
## Residual standard error: 4.085 on 69 degrees of freedom
## Multiple R-squared: 0.1908, Adjusted R-squared: 0.1439
## F-statistic: 4.067 on 4 and 69 DF, p-value: 0.005105
##
##
## ASSESSMENT OF THE LINEAR MODEL ASSUMPTIONS
## USING THE GLOBAL TEST ON 4 DEGREES-OF-FREEDOM:
## Level of Significance = 0.05
##
## Call:
## gvlma(x = lm(weight ~ gesttime + dose, data = litter))
##
## Value p-value Decision
## Global Stat 8.3233466 0.08043 Assumptions acceptable.
## Skewness 3.0816555 0.07918 Assumptions acceptable.
## Kurtosis 0.0002286 0.98794 Assumptions acceptable.
## Link Function 3.3202398 0.06843 Assumptions acceptable.
## Heteroscedasticity 1.9212226 0.16572 Assumptions acceptable.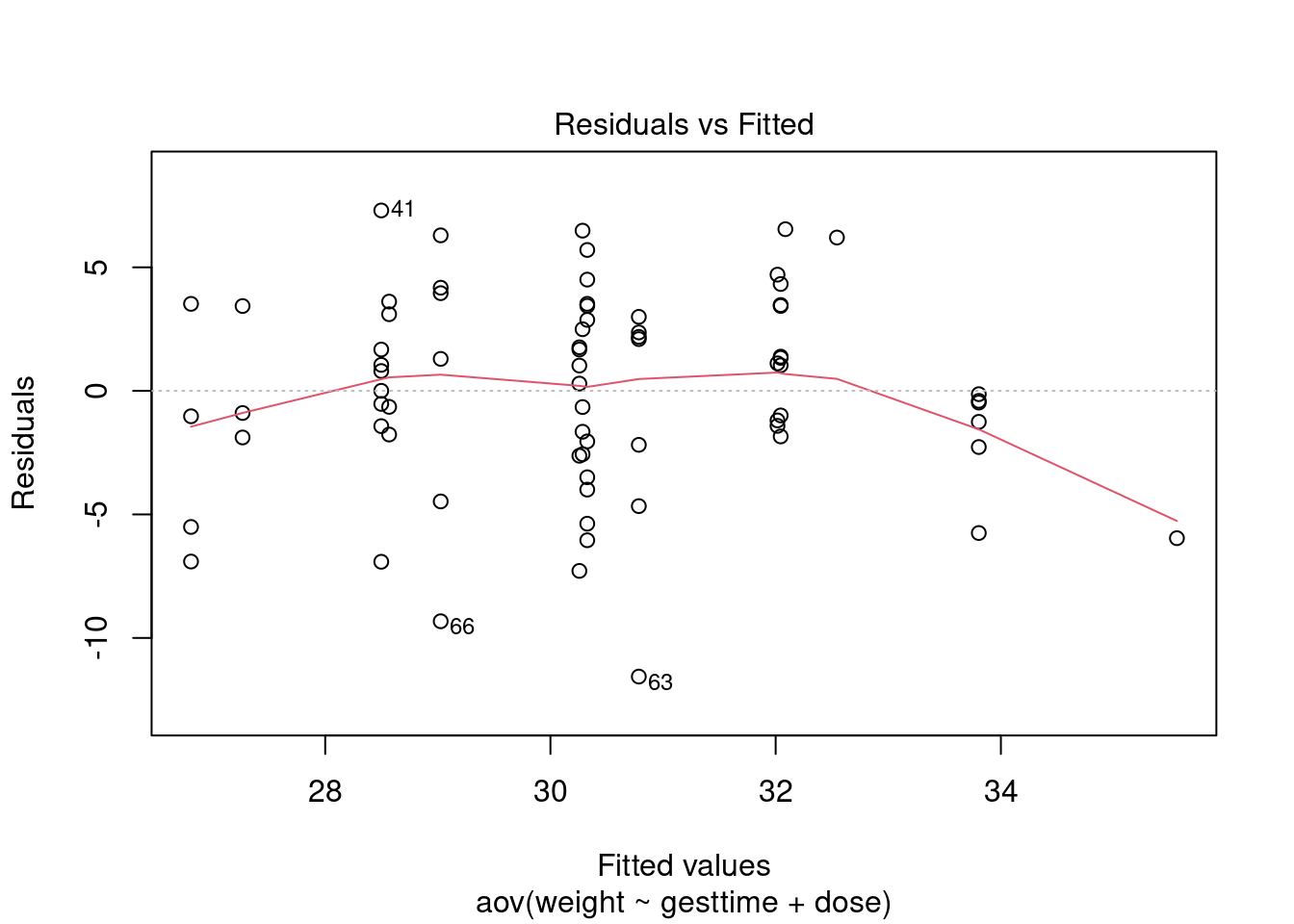
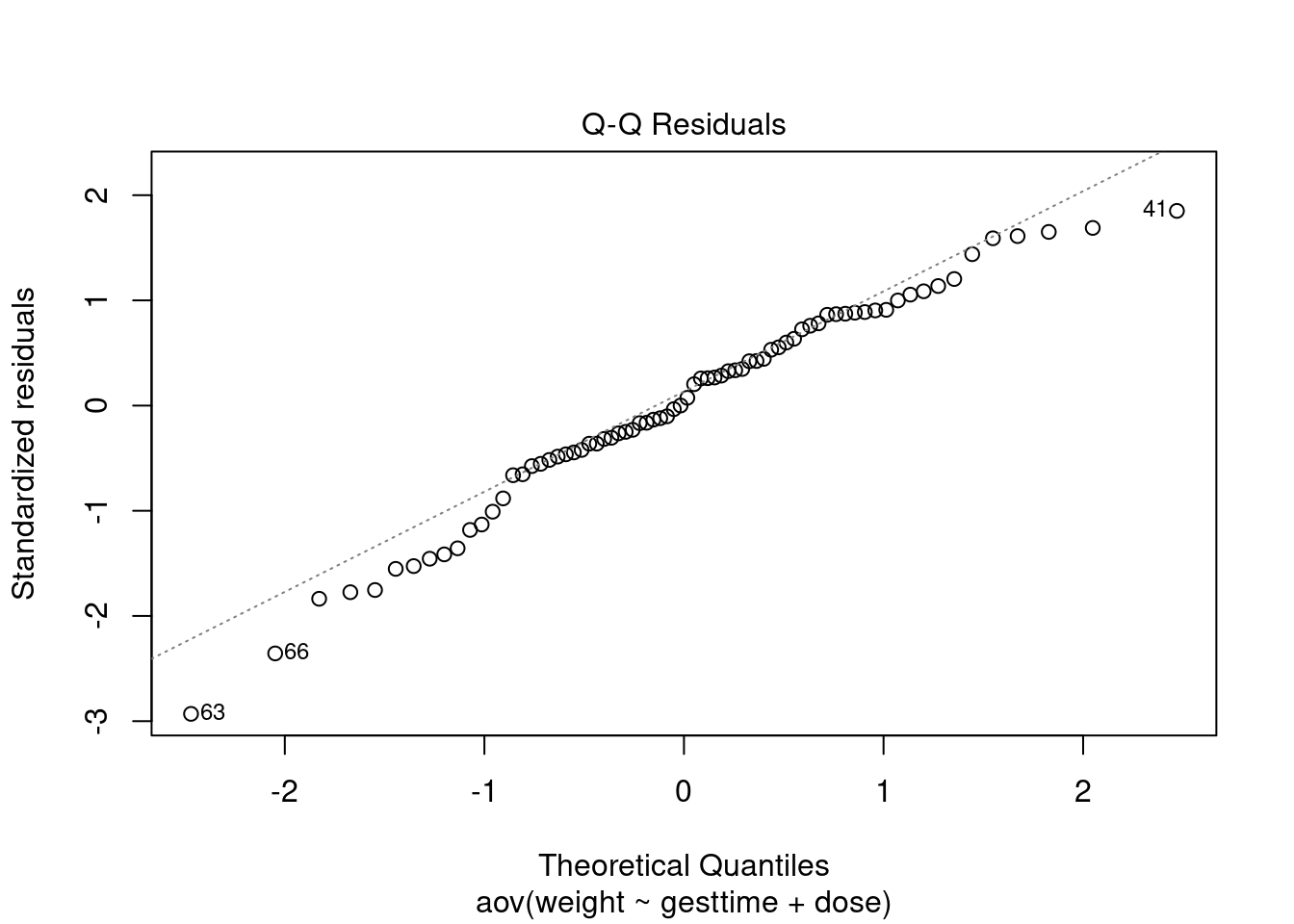
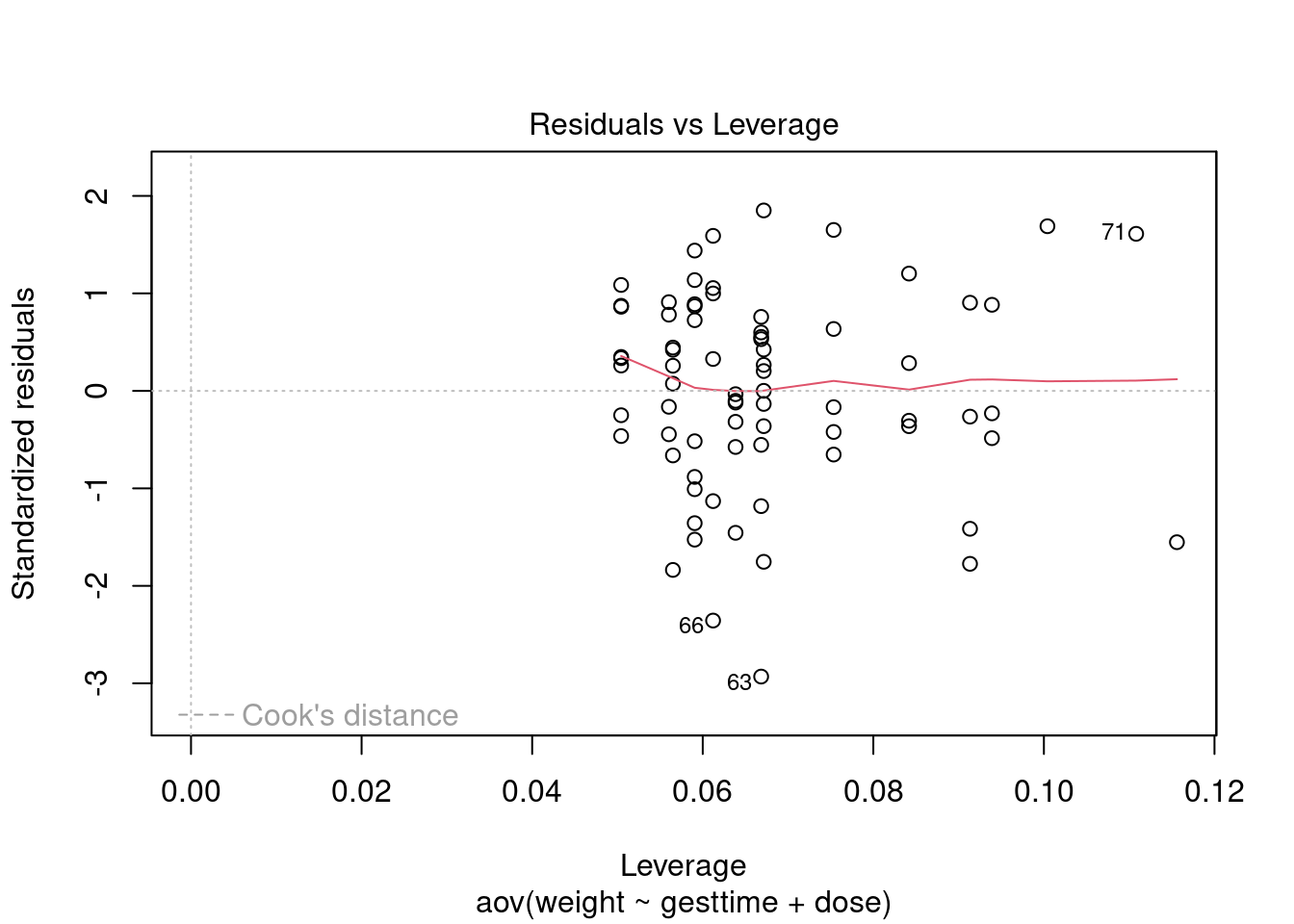
Normality
##
## Shapiro-Wilk normality test
##
## data: rstandard(litter_fit)
## W = 0.97617, p-value = 0.175Equality of variances
Note that the Brown-Forsythe test returns a significant result,
indicating heterogeneity of residual variances. However, this is for a
one-way model without the gesttime covariate. Including the
covariate accounts for significant residual variance and satisfies all
gvlma assumptions.
## Levene's Test for Homogeneity of Variance (center = median: litter)
## Df F value Pr(>F)
## group 3 3.1788 0.02925 *
## 70
## ---
## Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1MANOVA
MANOVA = Multivariate Analysis of Variance
MANOVA example
Ultrasound data
Import dataset
## [1] "US" "USC" "V" "VC" "Time"Perform multivariate ANOVA
## Df Pillai approx F num Df den Df Pr(>F)
## as.factor(Time) 2 0.65326 1.2127 8 20 0.3412
## Residuals 12## Df Wilks approx F num Df den Df Pr(>F)
## as.factor(Time) 2 0.35085 1.5486 8 18 0.2094
## Residuals 12## Df Hotelling-Lawley approx F num Df den Df Pr(>F)
## as.factor(Time) 2 1.8385 1.8385 8 16 0.1427
## Residuals 12## Df Roy approx F num Df den Df Pr(>F)
## as.factor(Time) 2 1.8321 4.5803 4 10 0.02324 *
## Residuals 12
## ---
## Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1Fisher’s exact
Fisher’s exact test arthritis example
Arthritis data from vcd package
Load dataset
## Loading required package: grid## ID Treatment Sex Age Improved
## Min. : 1.00 Placebo:43 Female:59 Min. :23.00 None :42
## 1st Qu.:21.75 Treated:41 Male :25 1st Qu.:46.00 Some :14
## Median :42.50 Median :57.00 Marked:28
## Mean :42.50 Mean :53.36
## 3rd Qu.:63.25 3rd Qu.:63.00
## Max. :84.00 Max. :74.00attach(Arthritis)
arthritis_table <- xtabs( ~ Treatment + Improved, data = Arthritis)
arthritis_table## Improved
## Treatment None Some Marked
## Placebo 29 7 7
## Treated 13 7 21Fisher’s exact test prostate cancer example
Prostate cancer incidence data from TRAMP Tomato Soy paper
Zuniga KE, Clinton SK, Erdman JW. The interactions of dietary tomato powder and soy germ on prostate carcinogenesis in the TRAMP model. Cancer Prev. Res. (2013). DOI: 10.1158/1940-6207.CAPR-12-0443
Create data frame
Data also available in fishers_exact_cpr_2013.csv
# tomato+soy vs control
diet <- c('tomsoy', 'tomsoy', 'control', 'control')
cancer <- c('cancer', 'nocancer', 'cancer', 'nocancer')
count <- c(12, 15, 29, 0)
PCaTomatoSoy <- data.frame(diet, cancer, count)
PCaTomatoSoy## diet cancer count
## 1 tomsoy cancer 12
## 2 tomsoy nocancer 15
## 3 control cancer 29
## 4 control nocancer 0Create contingency table from data frame
## cancer
## diet cancer nocancer
## control 29 0
## tomsoy 12 15Wilcoxon
Wilcoxon test example
UScrime dataset from MASS package
See R in Action section 7.5.1 for walkthrough.
Load dataset
## M So Ed Po1 Po2 LF M.F Pop NW U1 U2 GDP Ineq Prob Time y
## 1 151 1 91 58 56 510 950 33 301 108 41 394 261 0.084602 26.2011 791
## 2 143 0 113 103 95 583 1012 13 102 96 36 557 194 0.029599 25.2999 1635
## 3 142 1 89 45 44 533 969 18 219 94 33 318 250 0.083401 24.3006 578
## 4 136 0 121 149 141 577 994 157 80 102 39 673 167 0.015801 29.9012 1969
## 5 141 0 121 109 101 591 985 18 30 91 20 578 174 0.041399 21.2998 1234
## 6 121 0 110 118 115 547 964 25 44 84 29 689 126 0.034201 20.9995 682## So: 0
## [1] 0.038201
## ------------------------------------------------------------
## So: 1
## [1] 0.055552Perform Wilcoxon test
wilcox.test(
Prob ~ So,
data = UScrime,
paired = FALSE,
alternative = 'two.sided',
var.equal = TRUE,
conf.level = 0.95
)##
## Wilcoxon rank sum exact test
##
## data: Prob by So
## W = 81, p-value = 8.488e-05
## alternative hypothesis: true location shift is not equal to 0Test assumptions
Equality of variances
Note that, although the Wilcoxon rank sum test does not require normality, it does require homogeneity of variances and independence of random samples.
##
## F test to compare two variances
##
## data: Prob by So
## F = 0.624, num df = 30, denom df = 15, p-value = 0.2646
## alternative hypothesis: true ratio of variances is not equal to 1
## 95 percent confidence interval:
## 0.2360299 1.4396653
## sample estimates:
## ratio of variances
## 0.6240006Kruskal-Wallis
Kruskal-Wallis example
Cardiac magnesium levels from Smith BW J Food Res 2013 (doi: 10.5539/jfr.v2n1p168).
Import dataset
- In RStudio, tools->import dataset can also be used.
- Dataset is attached so variables can be named without reference to the original dataset.
Perform Kruskal-Wallis test
# Convert independent variable to a factor for use later by PMCMRplus
cardiacMg$diet = as.factor(cardiacMg$diet)
# Perform Kruskal-Wallis test
kruskal.test(mg ~ diet, data = cardiacMg)##
## Kruskal-Wallis rank sum test
##
## data: mg by diet
## Kruskal-Wallis chi-squared = 6.7757, df = 2, p-value = 0.03378Multiple comparisons
- Pairwise Wilcoxon testing with experiment-wide error correction
- Nemenyi test (pairwise test for multiple comparisons of mean rank sums). Not corrected for multiple comparisons.
- Dunnett non-parametric multiple comparisons
## Warning in wilcox.test.default(xi, xj, paired = paired, ...): cannot compute
## exact p-value with ties
## Warning in wilcox.test.default(xi, xj, paired = paired, ...): cannot compute
## exact p-value with ties
## Warning in wilcox.test.default(xi, xj, paired = paired, ...): cannot compute
## exact p-value with ties##
## Pairwise comparisons using Wilcoxon rank sum test with continuity correction
##
## data: cardiacMg$mg and cardiacMg$diet
##
## 0.1 0.4
## 0.4 0.885 -
## chow 0.063 0.048
##
## P value adjustment method: holm## Warning in kwAllPairsNemenyiTest.default(cardiacMg$mg, cardiacMg$diet, method =
## "Tukey"): Ties are present, p-values are not corrected.##
## Pairwise comparisons using Tukey-Kramer-Nemenyi all-pairs test with Tukey-Dist approximation## data: cardiacMg$mg and cardiacMg$diet## 0.1 0.4
## 0.4 0.992 -
## chow 0.039 0.044##
## P value adjustment method: single-step## alternative hypothesis: two.sided# Dunnett
library(nparcomp)
nparcomp(mg ~ diet,
data = cardiacMg,
type = 'Dunnett',
control = 'chow')##
## #------Nonparametric Multiple Comparisons for relative contrast effects-----#
##
## - Alternative Hypothesis: True relative contrast effect p is not equal to 1/2
## - Type of Contrast : Dunnett
## - Confidence level: 95 %
## - Method = Logit - Transformation
## - Estimation Method: Pairwise rankings
##
## #---------------------------Interpretation----------------------------------#
## p(a,b) > 1/2 : b tends to be larger than a
## #---------------------------------------------------------------------------#
## ## $Data.Info
## Sample Size
## 1 0.1 8
## 2 0.4 9
## 3 chow 3
##
## $Contrast
## 0.1 0.4 chow
## 0.1 - chow 1 0 -1
## 0.4 - chow 0 1 -1
##
## $Analysis
## Comparison Estimator Lower Upper Statistic p.Value
## 1 p( chow , 0.1 ) 0.042 0.002 0.541 -2.124749 0.06608568
## 2 p( chow , 0.4 ) 0.001 0.000 0.005 -9.757859 0.00000000
##
## $Overall
## Quantile p.Value
## 1 2.236422 0
##
## $input
## $input$formula
## mg ~ diet
##
## $input$data
## diet mg
## 1 chow 0.199
## 2 chow 0.363
## 3 chow 0.363
## 4 0.1 0.321
## 5 0.1 0.141
## 6 0.1 0.144
## 7 0.1 0.145
## 8 0.1 0.140
## 9 0.1 0.157
## 10 0.1 0.137
## 11 0.1 0.144
## 12 0.4 0.140
## 13 0.4 0.160
## 14 0.4 0.138
## 15 0.4 0.145
## 16 0.4 0.150
## 17 0.4 0.146
## 18 0.4 0.144
## 19 0.4 0.149
## 20 0.4 0.139
##
## $input$type
## [1] "Dunnett"
##
## $input$conf.level
## [1] 0.95
##
## $input$alternative
## [1] "two.sided" "less" "greater"
##
## $input$asy.method
## [1] "logit" "probit" "normal" "mult.t"
##
## $input$plot.simci
## [1] FALSE
##
## $input$control
## [1] "chow"
##
## $input$info
## [1] TRUE
##
## $input$rounds
## [1] 3
##
## $input$contrast.matrix
## NULL
##
## $input$correlation
## [1] FALSE
##
## $input$weight.matrix
## [1] FALSE
##
##
## $text.Output
## [1] "True relative contrast effect p is not equal to 1/2"
##
## $connames
## [1] "p( chow , 0.1 )" "p( chow , 0.4 )"
##
## $AsyMethod
## [1] "Logit - Transformation"
##
## attr(,"class")
## [1] "nparcomp"Test assumptions
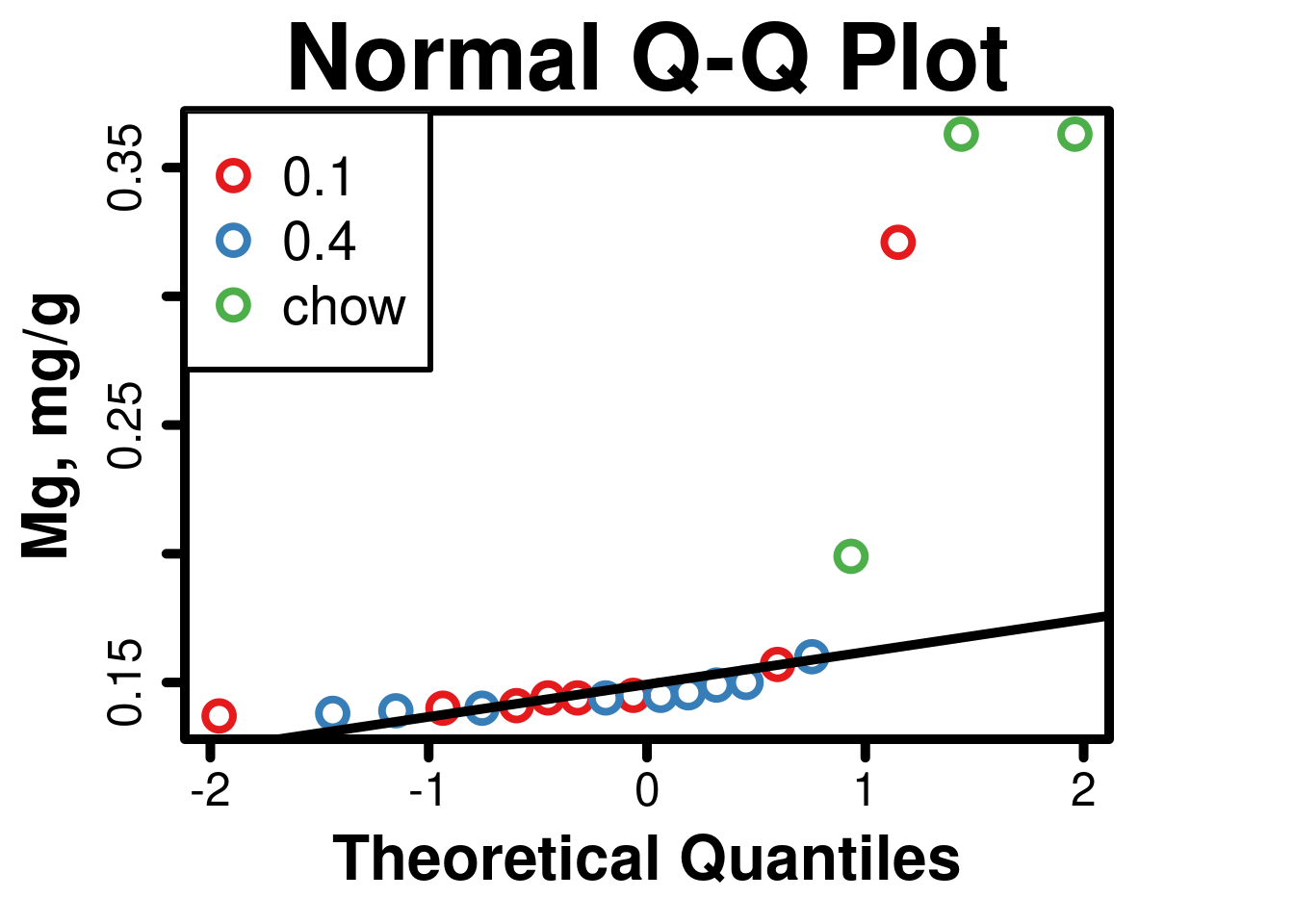
Normality
- The RColorBrewer package is loaded for additional colors, and a
palette is added to R. Use brackets after vector name for specific
colors when creating plots (for example:
col=set1[4:14]). - Legend can also be manually positioned with xy coordinates (numbers from 0,0 at bottom left of plot to 1,1 at top right)
- As demonstrated by the tests below, these data fail the assumption of normality, and are not amenable to transformation, hence the use of non-parametric testing.
##
## Shapiro-Wilk normality test
##
## data: cardiacMg$mg
## W = 0.55747, p-value = 1.101e-06# Normal Quantile-Quantile Plot
# Set graphical parameters
par(
cex = 1,
cex.main = 3,
cex.lab = 2,
font.lab = 2,
cex.axis = 1.5,
lwd = 5,
lend = 0,
ljoin = 0,
tcl = -0.5,
pch = 1,
mar = c(5, 5, 3, 5)
)
# Load RColorBrewer and set palette
library(RColorBrewer)
set1 <- brewer.pal(8, 'Set1')
# Create vector for categories
categories <- factor(cardiacMg$diet)
# Create plot
qqnorm(
mg,
ylab = 'Mg, mg/g',
axes = FALSE,
col = set1[categories],
lwd = 4,
cex = 2
)
# Add normal line
qqline(cardiacMg$mg, lwd = 5)
# Add border to complete axes
box(which = 'plot', bty = 'o', lwd = 5)
axis(1, lwd = 5, tcl = -0.5) # x axis
axis(2, lwd = 5, tcl = -0.5) # y axis
# Add legend
legend(
'topleft',
legend = levels(categories),
box.lwd = 3,
pch = 1.5,
cex = 1.75,
pt.cex = 2,
pt.lwd = 4,
col = set1
)
Equality of variances
- This is a brown-forsythe test that evaluates deviations from group medians, and thus is robust to violations of normality.
- Fligner test included for comparison.
##
## Modified robust Brown-Forsythe Levene-type test based on the absolute
## deviations from the median
##
## data: cardiacMg$mg
## Test Statistic = 1.1301, p-value = 0.3461##
## Fligner-Killeen test of homogeneity of variances
##
## data: cardiacMg$mg by cardiacMg$diet
## Fligner-Killeen:med chi-squared = 0.11837, df = 2, p-value = 0.9425Generate boxplot
- Parameters (
par): Graphical parameters need to be set again because of code chunk separation in RMarkdown. Normally they would only be set once per R session. - X axis: The default axes will usually detect
categorical data and label axes accordingly, but categories will need to
be specified for axes created separately. It is easiest to create a
separate vector with the categories, and then reference it when creating
the plot. Labels can also be specified with
labels=c('group1', 'group2'). - Y axis: Note that it does not always start at zero, but encompasses the data range with some padding.
# Set graphical parameters
par(
cex = 1,
cex.main = 3,
cex.lab = 2,
font.lab = 2,
cex.axis = 1.5,
lwd = 5,
lend = 0,
ljoin = 0,
tcl = -0.5,
pch = 1,
mar = c(5, 5, 3, 5)
)
# Create boxplot
boxplot(
cardiacMg$mg ~ cardiacMg$diet,
main = 'Cardiac mg',
xlab = 'diet',
ylab = 'Mg, mg/g',
axes = FALSE,
lwd = 4,
medlwd = 7,
outlwd = 4,
cex = 2,
col = set1
)
# Add border to complete axes
box(which = 'plot', bty = 'l', lwd = 5)
# Create vector with means of each group
means <- aggregate(mg ~ diet, data = cardiacMg, mean)
# Add points for group means to boxplot
points(
means$mg,
cex = 2,
pch = 23,
bg = 0,
lwd = 4
)
# X axis
axis(
1,
lwd = 5,
tcl = -0.5,
labels = levels(categories),
at = c(1:3)
)
# Y axis
axis(2, lwd = 5, tcl = -0.5)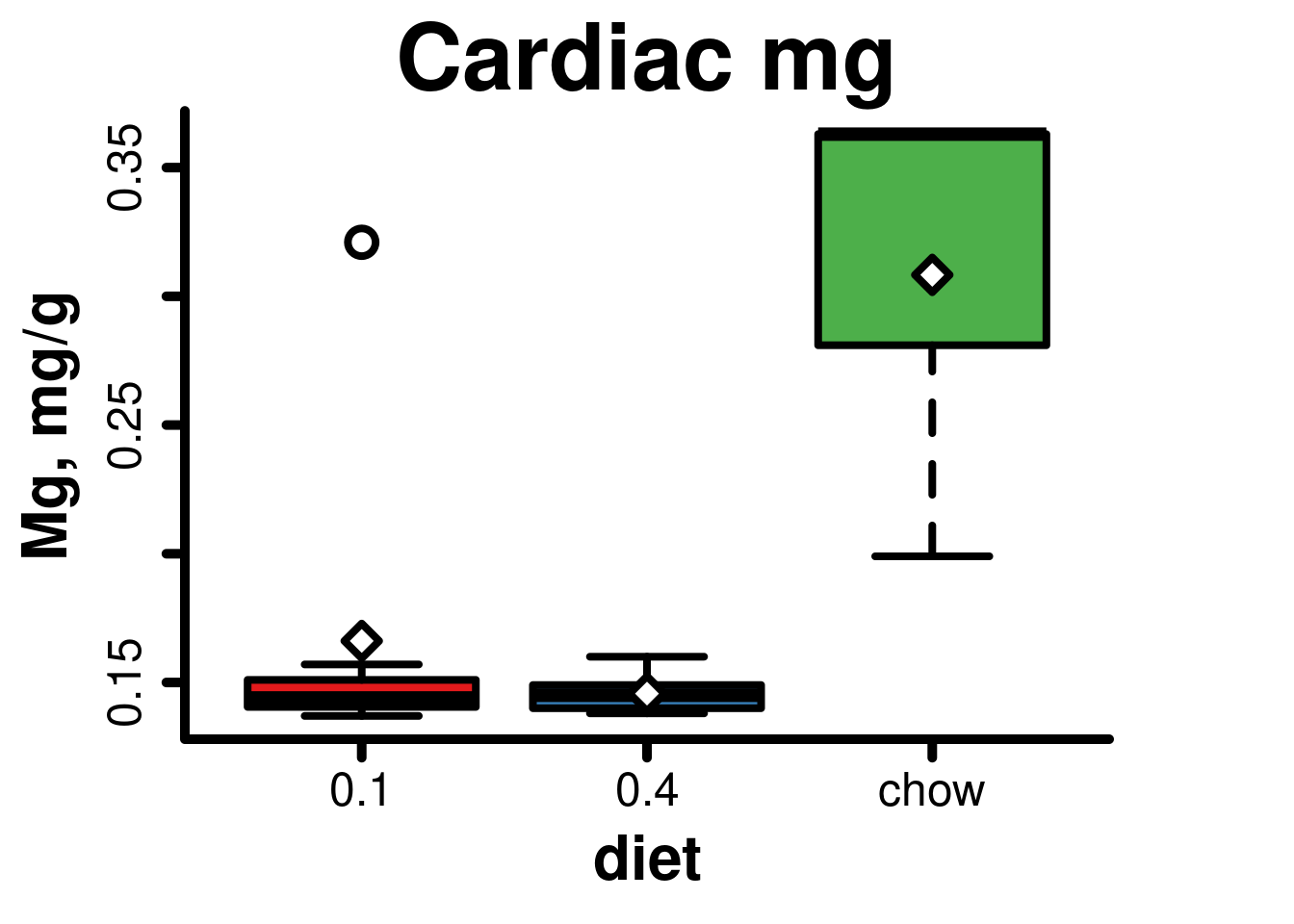
Plots
General examples
ggplot2 qplot example
Example from ggplot2 documentation p. 135
Intro
This file demonstrates some of the capabilities in the ggplot2
package. The mtcars dataset is included with R. The beginning of the
dataset is shown with head(mtcars) to verify.
Import data
## mpg cyl disp hp drat wt qsec vs am gear carb
## Mazda RX4 21.0 6 160 110 3.90 2.620 16.46 0 1 4 4
## Mazda RX4 Wag 21.0 6 160 110 3.90 2.875 17.02 0 1 4 4
## Datsun 710 22.8 4 108 93 3.85 2.320 18.61 1 1 4 1
## Hornet 4 Drive 21.4 6 258 110 3.08 3.215 19.44 1 0 3 1
## Hornet Sportabout 18.7 8 360 175 3.15 3.440 17.02 0 0 3 2
## Valiant 18.1 6 225 105 2.76 3.460 20.22 1 0 3 1Plot data
## Warning: `qplot()` was deprecated in ggplot2 3.4.0.
## This warning is displayed once every 8 hours.
## Call `lifecycle::last_lifecycle_warnings()` to see where this warning was
## generated.
# It will use data from local environment
hp <- mtcars$hp
wt <- mtcars$wt
cyl <- mtcars$cyl
vs <- mtcars$vs
am <- mtcars$am
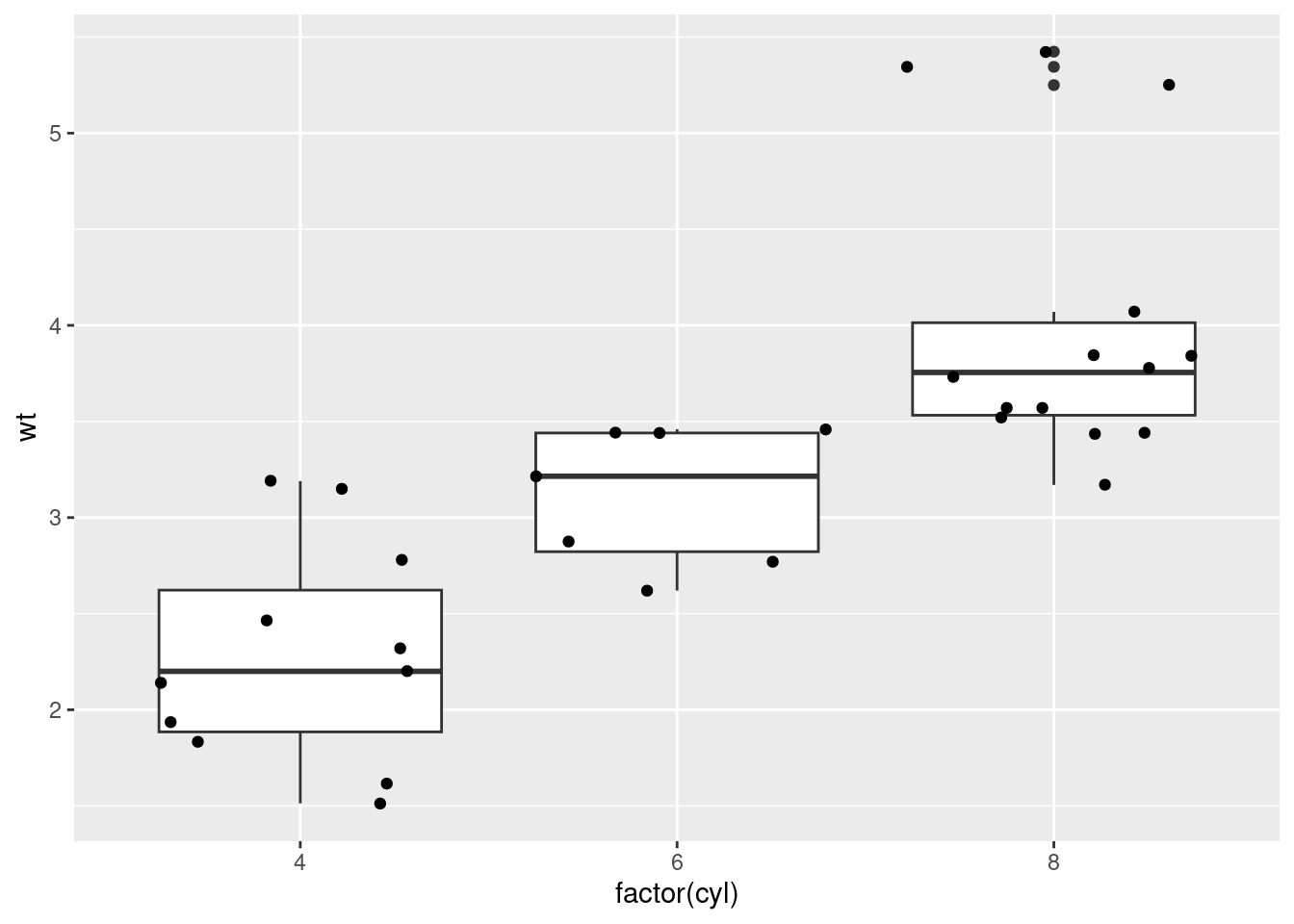
qplot(hp, wt)
# qplot will attempt to guess what geom you want depending on the input
# both x and y supplied = scatterplot
qplot(mpg, wt, data = mtcars)
## `stat_bin()` using `bins = 30`. Pick better value with `binwidth`.
## Bin width defaults to 1/30 of the range of the data. Pick better value with
## `binwidth`.
Creating rotated axis labels in R
7.27 How can I create rotated axis labels?
Intro
To rotate axis labels (using base graphics), you need to use text(), rather than mtext(), as the latter does not support par(‘srt’).
Example plot
# Increase bottom margin to make room for rotated labels
par(mar = c(7, 4, 4, 2) + 0.1)
# Create plot with no x axis and no x axis label
plot(1:8, xaxt = 'n', xlab = '')
# Set up x axis with tick marks alone
axis(1, labels = FALSE)
# Create some text labels
labels <- paste('Label', 1:8, sep = ' ')
# Plot x axis labels at default tick marks
text(
1:8,
par('usr')[3] - 0.25,
srt = 45,
adj = 1,
labels = labels,
xpd = TRUE
)
# Plot x axis label at line 6 (of 7)
mtext(1, text = 'X Axis Label', line = 6)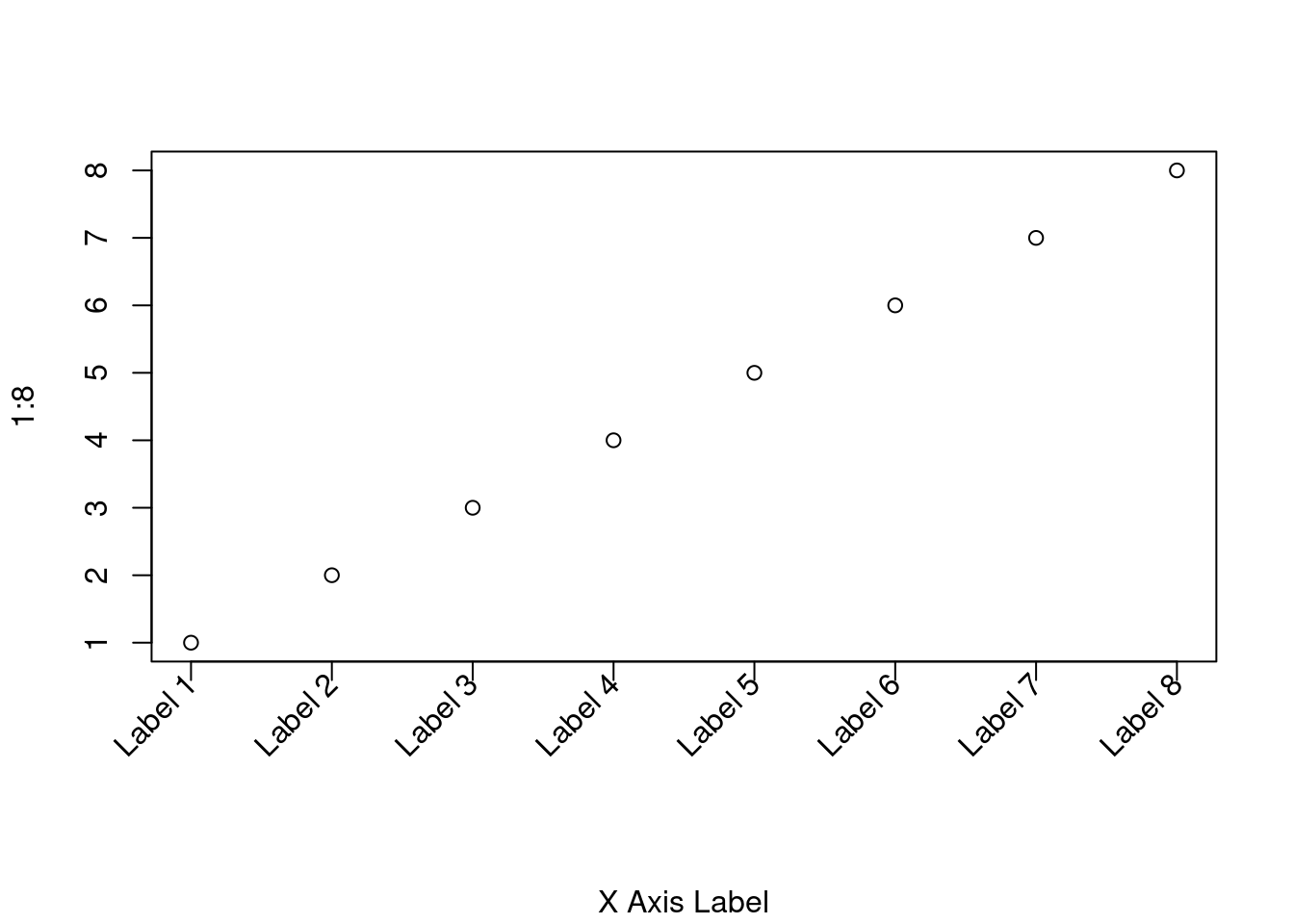
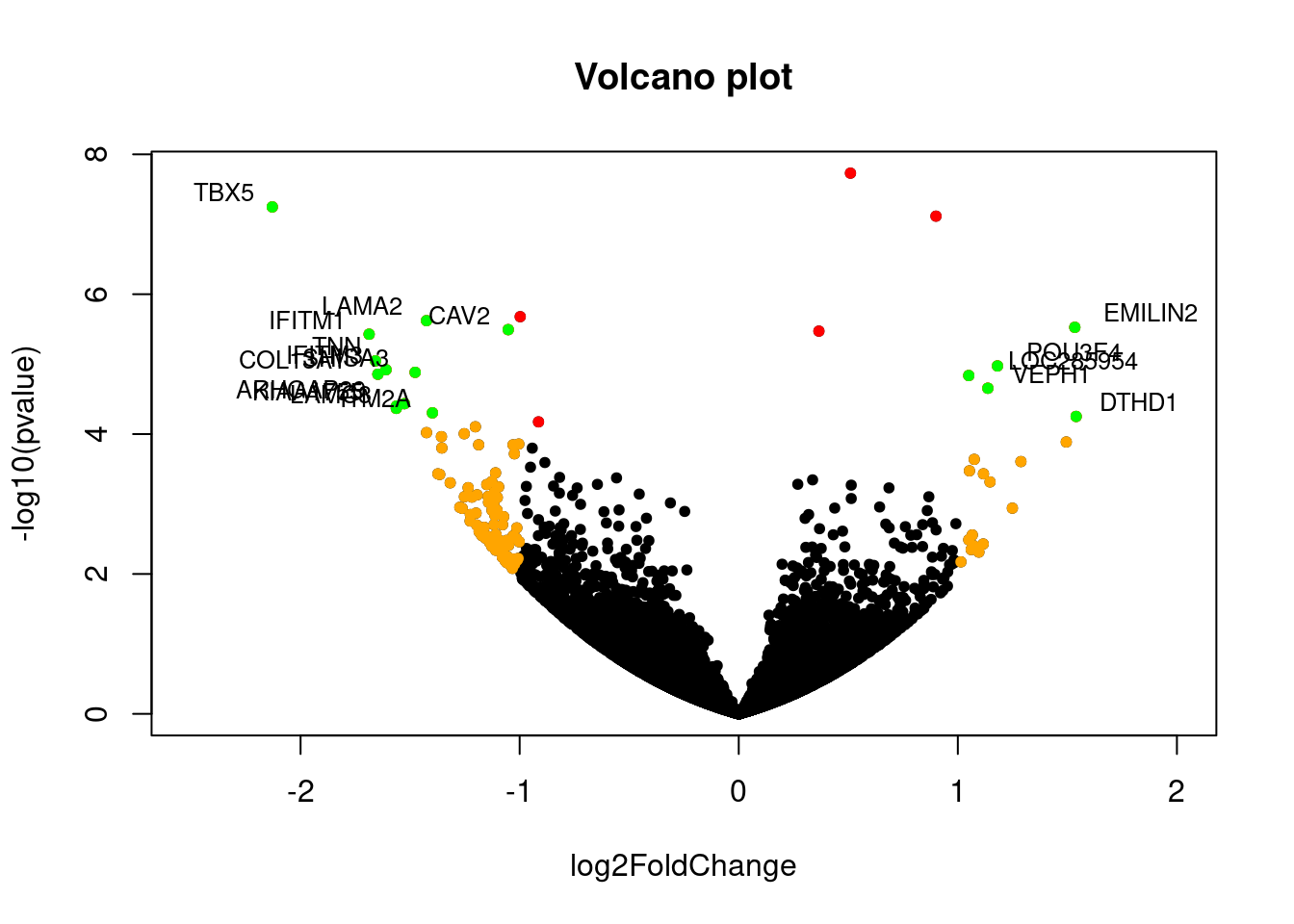
Volcano plots
Getting Genetics Done example
Volcano plot example from Getting Genetics Done, with data from this GitHub Gist.
Import dataset from GitHub
## Gene log2FoldChange pvalue padj
## 1 DOK6 0.5100 1.861e-08 0.0003053
## 2 TBX5 -2.1290 5.655e-08 0.0004191
## 3 SLC32A1 0.9003 7.664e-08 0.0004191
## 4 IFITM1 -1.6870 3.735e-06 0.0068090
## 5 NUP93 0.3659 3.373e-06 0.0068090
## 6 EMILIN2 1.5340 2.976e-06 0.0068090Make a basic volcano plot
The -log10(pvalue) is used so p values can be plotted as
whole numbers. Note that I was not able to separate each section into
different R markdown code chunks. I needed the generation of points to
be continuous with generation of the original plot.
with(res,
plot(
log2FoldChange,
-log10(pvalue),
pch = 20,
main = 'Volcano plot',
xlim = c(-2.5, 2)
))
# Add colored points:
# red if padj<0.05
# orange if log2FC>1
# green if both
with(subset(res, padj < .05),
points(
log2FoldChange,
-log10(pvalue),
pch = 20,
col = 'red'
))
with(subset(res, abs(log2FoldChange) > 1),
points(
log2FoldChange,
-log10(pvalue),
pch = 20,
col = 'orange'
))
with(subset(res, padj < .05 & abs(log2FoldChange) > 1),
points(
log2FoldChange,
-log10(pvalue),
pch = 20,
col = 'green'
))
# Label points with the textxy function from the calibrate package
library(calibrate)
with(subset(res, padj < .05 & abs(log2FoldChange) > 1),
textxy(
log2FoldChange,
-log10(pvalue),
labs = Gene,
cex = .8
))
Nrf1 proteomics volcano plot
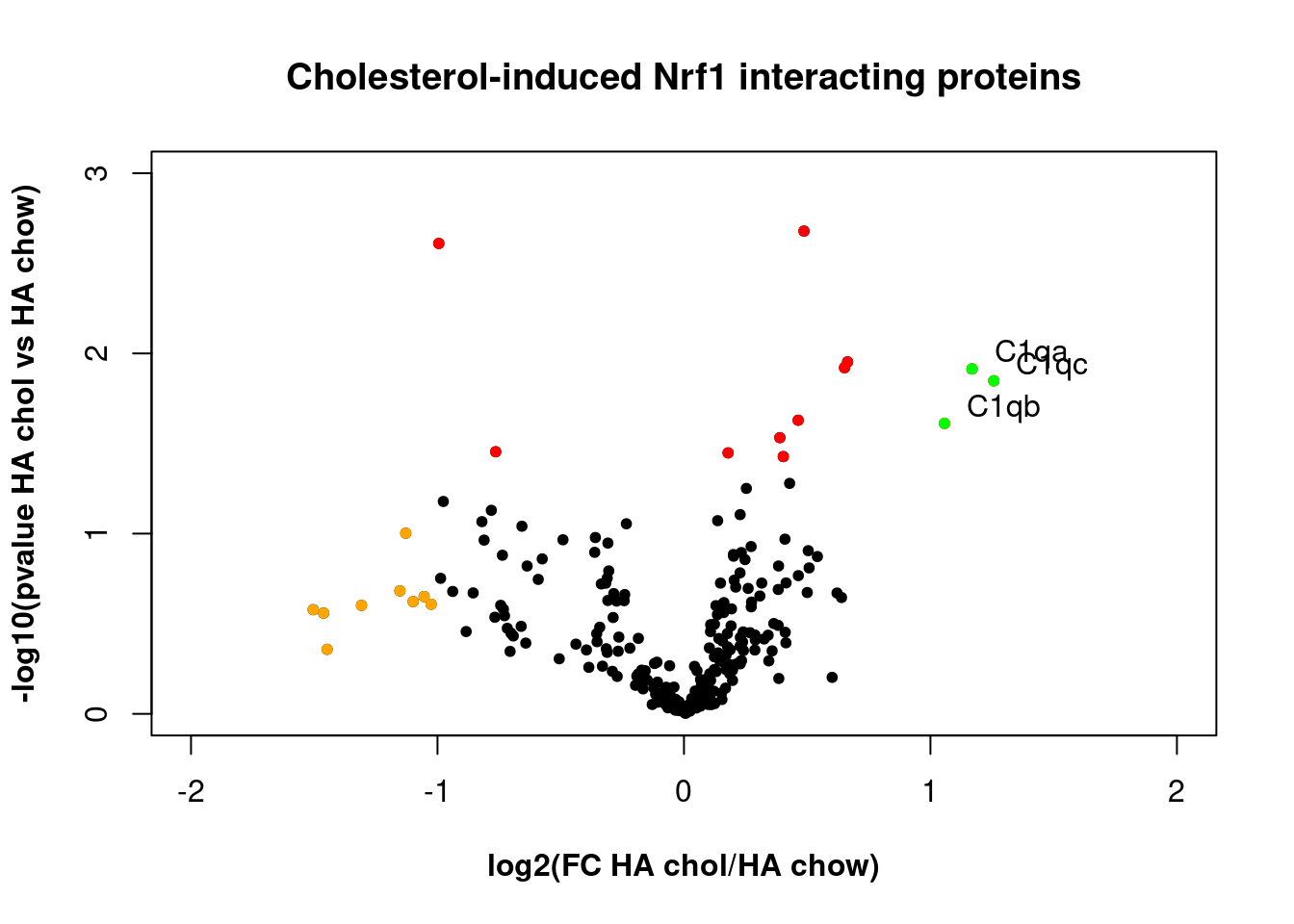
Experimental data example from Nrf1 proteomics project. The goal of this experiment was to identify a molecular complex associated with Nrf1, a protein molecule our research group was studying. Complement C1q proteins were identified as potentially interacting with Nrf1.
Import data
proteomics_data <-
read.table(
'https://raw.githubusercontent.com/br3ndonland/R-proteomics-Nrf1/master/data/R-proteomics-Nrf1.csv',
header = TRUE,
sep = ','
)
attach(proteomics_data)
head(proteomics_data)## Gene log2_FC_HAchol_HAchow log2_FC_HAchow_lacZ pvalue pvaluechow
## 1 Serpinb5 0.09538888 -1.3634997 0.74081917 0.39977101
## 2 Sfn 0.41354494 -1.0428302 0.40347276 0.20906780
## 3 Eno1 0.63929006 -0.6662456 0.22617602 0.32692726
## 4 C1qc 1.25725953 0.0891710 0.01419030 0.86011207
## 5 C1qa 1.16884182 0.0303050 0.01219113 0.95321354
## 6 Gstm1 0.19700734 -0.8650671 0.65225515 0.09657287
## log2_FC_HAbort_HAchow pvaluebort
## 1 1.7303640 0.31471162
## 2 2.5999482 0.01377441
## 3 0.8034121 0.06946092
## 4 0.0946041 0.84034899
## 5 0.1015774 0.80231428
## 6 0.4450431 0.21202782Generate plots
- Each point is a protein.
- Red if p<0.05 for the comparison shown
- Orange if [log2 fold change]>1
- Green if both
- log2 transformation is used to normalize positive and negative fold changes.
- -log10(pvalue) is used so p values can be plotted as whole numbers.
HA cholesterol plot
This plot compares chow-fed mice with mice fed the Paigen diet to promote accumulation of cholesterol in the liver, and demonstrates the increased abundance of Complement C1qA, C1qB, and C1qC, in mice fed the Paigen diet.
# bold axis titles
par(font.lab = 2)
# create plot
with(
proteomics_data,
plot(
log2_FC_HAchol_HAchow,
-log10(pvalue),
pch = 20,
xlim = c(-2, 2),
ylim = c(0, 3),
yaxp = c(0, 3, 3),
main = 'Cholesterol-induced Nrf1 interacting proteins',
xlab = 'log2(FC HA chol/HA chow)',
ylab = '-log10(pvalue HA chol vs HA chow)'
)
)
# Add colored points:
# red if pvalue<0.05, orange if [log2FC]>1, green if both)
with(
subset(proteomics_data, pvalue < .05),
points(
log2_FC_HAchol_HAchow,
-log10(pvalue),
pch = 20,
col = 'red'
)
)
with(
subset(proteomics_data, abs(log2_FC_HAchol_HAchow) > 1),
points(
log2_FC_HAchol_HAchow,
-log10(pvalue),
pch = 20,
col = 'orange'
)
)
with(
subset(proteomics_data, pvalue < .05 &
abs(log2_FC_HAchol_HAchow) > 1),
points(
log2_FC_HAchol_HAchow,
-log10(pvalue),
pch = 20,
col = 'green'
)
)
# Label points with the textxy function from the calibrate package
library(calibrate)
with(
subset(proteomics_data, pvalue < .05 &
abs(log2_FC_HAchol_HAchow) > 1),
textxy(
log2_FC_HAchol_HAchow,
-log10(pvalue),
labs = Gene,
cex = 1
)
)
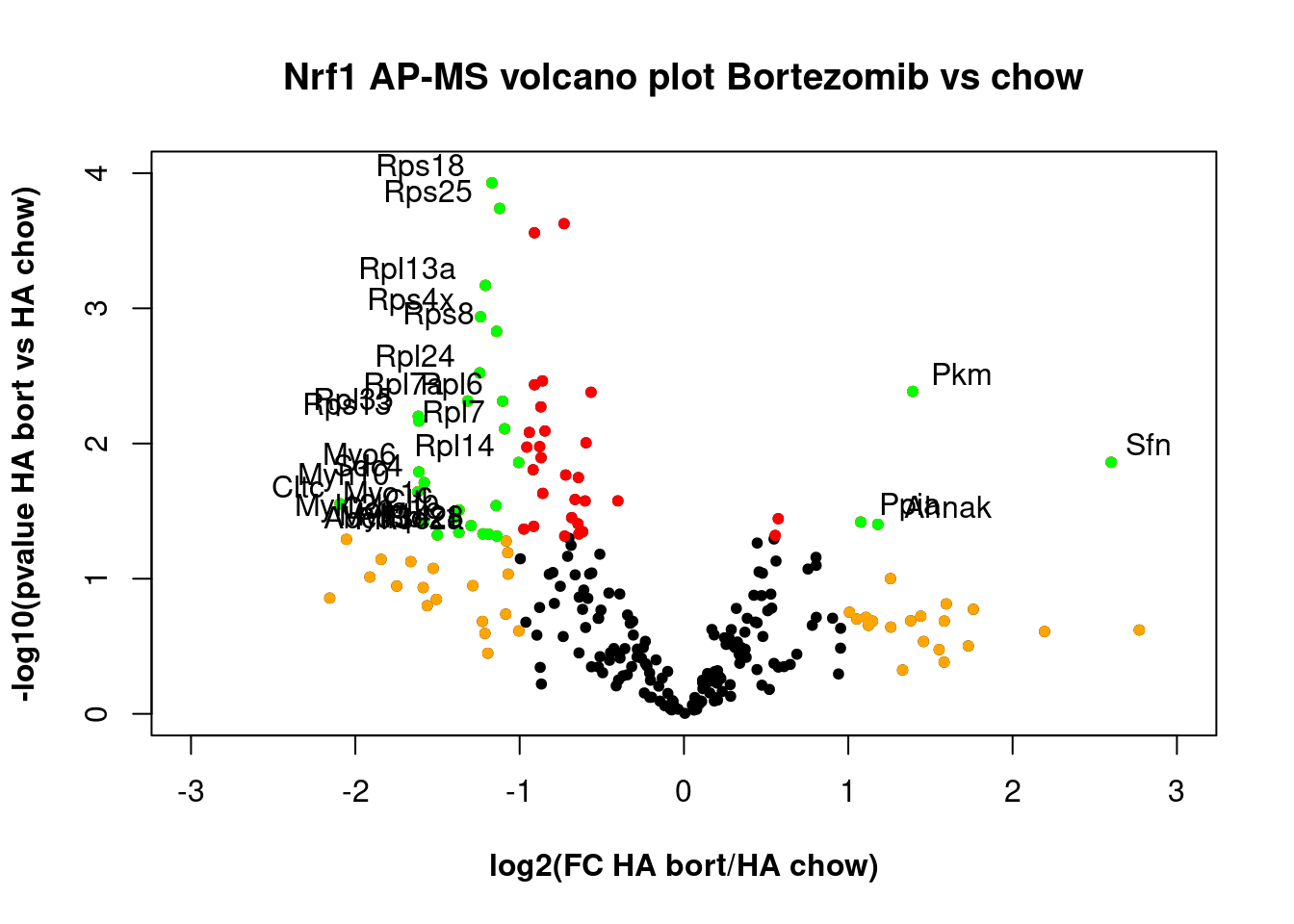
HA Bortezomib plot
Bortezomib treatment alters the proteome.
# bold axis titles
par(font.lab = 2)
# create plot
with(
proteomics_data,
plot(
log2_FC_HAbort_HAchow,
-log10(pvaluebort),
pch = 20,
xlim = c(-3, 3),
ylim = c(0, 4),
main = 'Nrf1 AP-MS volcano plot Bortezomib vs chow',
xlab = 'log2(FC HA bort/HA chow)',
ylab = '-log10(pvalue HA bort vs HA chow)'
)
)
# Add colored points:
# red if pvalue<0.05, orange if log2FC>1, green if both)
with(
subset(proteomics_data, pvaluebort < .05),
points(
log2_FC_HAbort_HAchow,
-log10(pvaluebort),
pch = 20,
col = 'red'
)
)
with(
subset(proteomics_data, abs(log2_FC_HAbort_HAchow) > 1),
points(
log2_FC_HAbort_HAchow,
-log10(pvaluebort),
pch = 20,
col = 'orange'
)
)
with(
subset(
proteomics_data,
pvaluebort < .05 & abs(log2_FC_HAbort_HAchow) > 1
),
points(
log2_FC_HAbort_HAchow,
-log10(pvaluebort),
pch = 20,
col = 'green'
)
)
# Label points with the textxy function from the calibrate package
library(calibrate)
with(
subset(
proteomics_data,
pvaluebort < .05 & abs(log2_FC_HAbort_HAchow) > 1
),
textxy(
log2_FC_HAbort_HAchow,
-log10(pvaluebort),
labs = Gene,
cex = 1
)
)
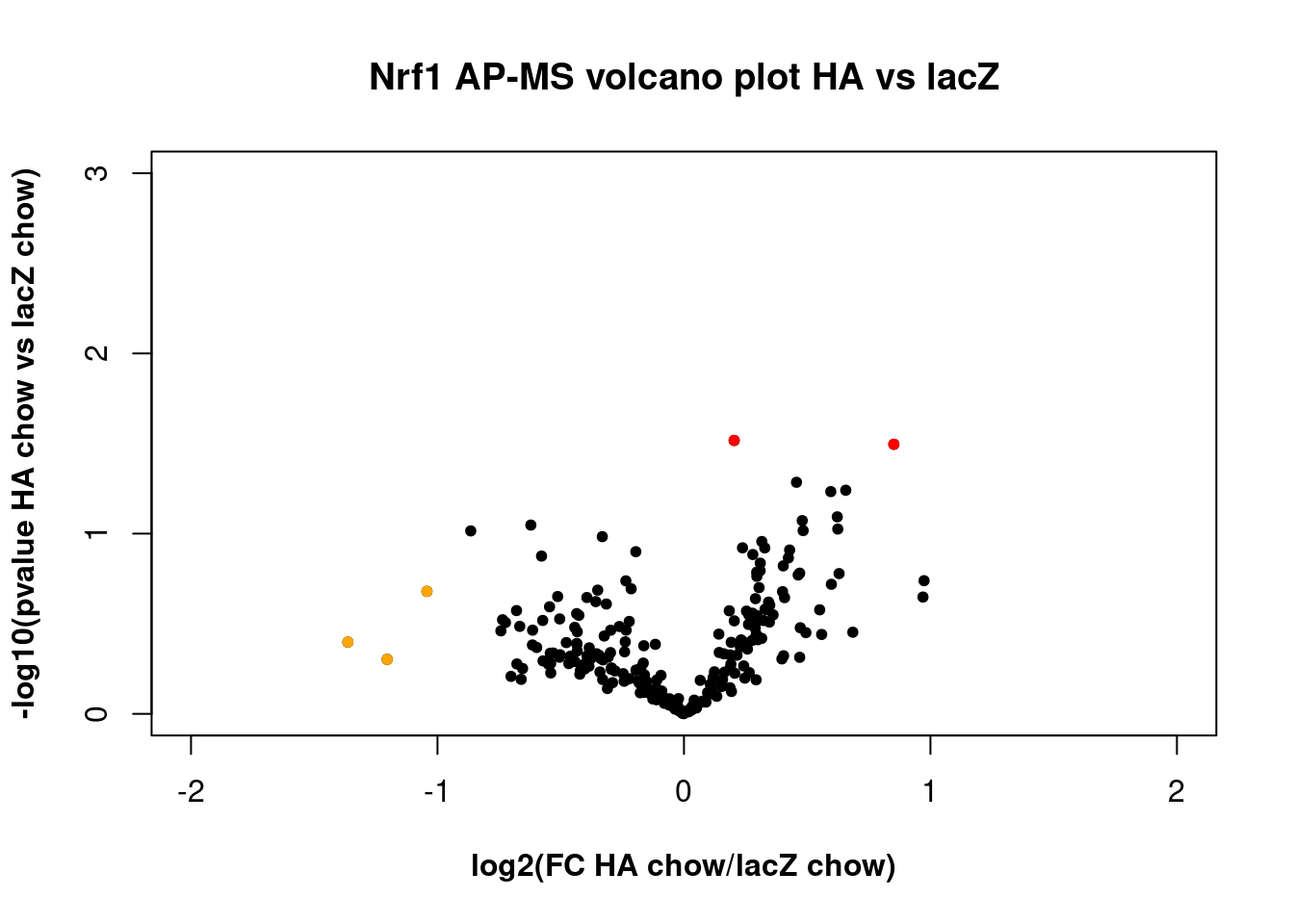
HA vs lacZ plot
The proteome was not significantly different with or without the HA tag, indicating issues with the HA immunoprecipitation. Note that the code for labeling points appears to be separated from the rest of the code for the plot, possibly because there were no points labeled.
# bold axis titles
par(font.lab = 2)
# create plot
with(
proteomics_data,
plot(
log2_FC_HAchow_lacZ,-log10(pvaluechow),
pch = 20,
xlim = c(-2, 2),
ylim = c(0, 3),
yaxp = c(0, 3, 3),
main = 'Nrf1 AP-MS volcano plot HA vs lacZ',
xlab = 'log2(FC HA chow/lacZ chow)',
ylab = '-log10(pvalue HA chow vs lacZ chow)'
)
)
# Add colored points:
# red if pvalue<0.05, orange if [log2FC]>1, green if both)
with(
subset(proteomics_data, pvaluechow < .05),
points(
log2_FC_HAchow_lacZ,-log10(pvaluechow),
pch = 20,
col = 'red'
)
)
with(
subset(proteomics_data, abs(log2_FC_HAchow_lacZ) > 1),
points(
log2_FC_HAchow_lacZ,-log10(pvaluechow),
pch = 20,
col = 'orange'
)
)
with(
subset(proteomics_data, pvaluechow < .05 &
abs(log2_FC_HAchow_lacZ) > 1),
points(
log2_FC_HAchow_lacZ,-log10(pvaluechow),
pch = 20,
col = 'green'
)
)
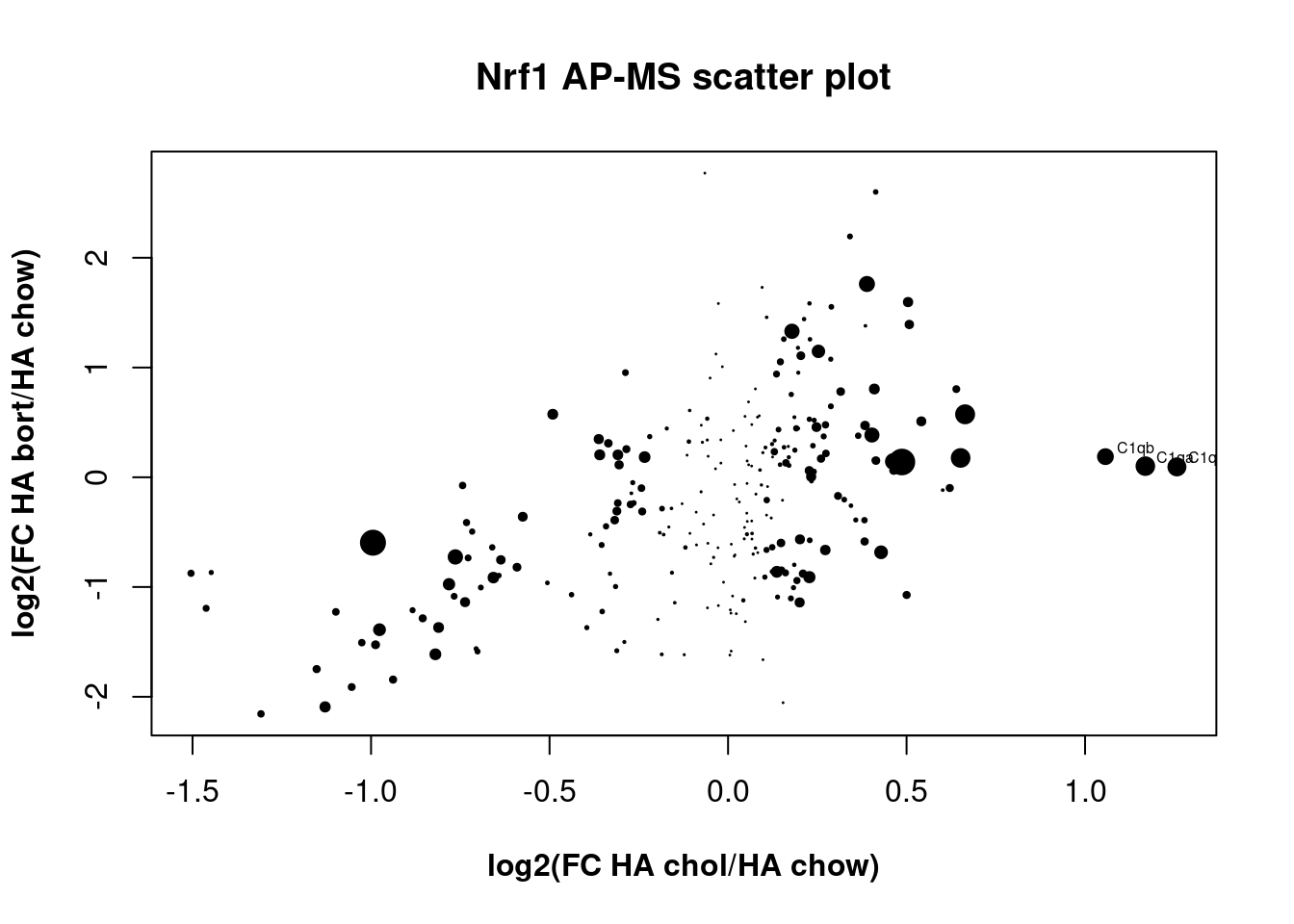
Scatter plot
The scatter plot is an interesting way to visualize two between-group comparisons, but the volcano plot is more useful for this dataset.
To set size of points according to p value:
dotsize <- -log10(pvalue)
head(dotsize)then put cex=dotsize inside the plot() and
points() functions.
# set size of points according to p value
dotsize <- -log10(pvalue)
# bold axis titles
par(font.lab = 2)
# create plot
with(
proteomics_data,
plot(
log2_FC_HAchol_HAchow,
log2_FC_HAbort_HAchow,
pch = 20,
main = 'Nrf1 AP-MS scatter plot',
xlab = 'log2(FC HA chol/HA chow)',
ylab = 'log2(FC HA bort/HA chow)',
cex = dotsize
)
)
# Label points with the textxy function from the calibrate package
library(calibrate)
with(
subset(proteomics_data, pvalue < .05 &
abs(log2_FC_HAchol_HAchow) > 1),
textxy(log2_FC_HAchol_HAchow,
log2_FC_HAbort_HAchow,
labs = Gene)
)
Comments